

1 Introduction
〚Pd(C3H5)Cl〛2 is often used as the palladium source in catalytic allylic substitution reactions, assuming that, in the presence of a bidentate ligand L2, the cationic active species Pd(C3H5)(L2)+ is readily formed with Cl– as a simple non-coordinating anion 〚1〛. However, it has been reported that halide ions, and particularly Cl–, can have an influence on the reactivity or the selectivity of Pd-catalysed 〚2–7〛 allylic substitution reactions. In some cases, it has been proposed that this influence is originated from the well-known increase of the isomerisation rate of the Pd η3-allyl intermediates through η3–η1–η3 interconversion 〚6–8〛. The working models of the key catalytic intermediate are mostly cationic palladium η3-allyl complexes with a non-coordinating anion, e.g. triflate. The study of these models has proven to be particularly useful in asymmetric allylic alkylation with chelating chiral ligands 〚1〛. If the anion is Cl–, which is the case after the reaction of 〚Pd(η3-allyl)Cl〛2 with 4 equiv monodentate ligand or 2 equiv bidentate ligand, the complex generally remains η3-allyl: a cationic tetracoordinate complex with a non-coordinating chloride anion, both being present either as separate ions or as tight ion pairs, or a neutral pentacoordinate chloro complex 〚8–13〛. However, neutral tetracoordinate palladium η1-allyl chloro complexes have also been reported 〚14, 15〛.
We have been interested recently in phospholane-derived ligands and their use in palladium-catalysed allylic alkylation 〚16, 17〛. We report hereafter our observations concerning the effect of chloride ions on the NMR characteristics and the chemical reactivity of some palladium(II) allyl complexes bearing the bis-phospholane ligands (R,R)-Me-Duxantphospholane or (R,R)-Me-Duphos, or the bis-phosphine ligand (S)-Binap.
2 Results and discussion
The palladium complexes we have studied by NMR spectroscopy are listed in Fig. 1. Adding a source of chloride ions (viz. 1 equiv tetraphenylphosphonium chloride) to a solution of 〚1〛SbF6 in deuterated chloroform induces a significant alteration of the allylic signals on the 1H NMR spectrum (Fig. 2). At room temperature, the central allylic proton (Hc) of 〚1〛SbF6 appears as a pseudo-triplet at 6.73 ppm, with an expected value of 13 Hz for 3JHH; the signals of the terminal allylic protons (Ht and Ht’) are degenerate at 5.20 ppm, with coupling constants with the phosphorus in trans position (3JHP) of ca 13 Hz (Fig. 2a). Upon addition of Cl–, the signals of the terminal allylic protons are strongly shifted to higher frequency at 5.46 and 6.04 ppm, whereas the chemical shift of the Hc signal is almost unchanged. However, the multiplicity and the coupling constants are globally unchanged; incidentally the value of 3JHP with the phosphorus cis (3.4 Hz) can be determined for the signal at 6.04 ppm. This shows that the allyl ligand remains η3-coordinated and Me-Duphos η2-coordinated on the palladium centre. In addition, both phosphorus atoms of Me-Duphos lie in the equatorial plane, by contrast with the nitrogens of neocuproine in the palladium chloro allyl complex described by Åkermark, Vitagliano et al. 〚8〛.
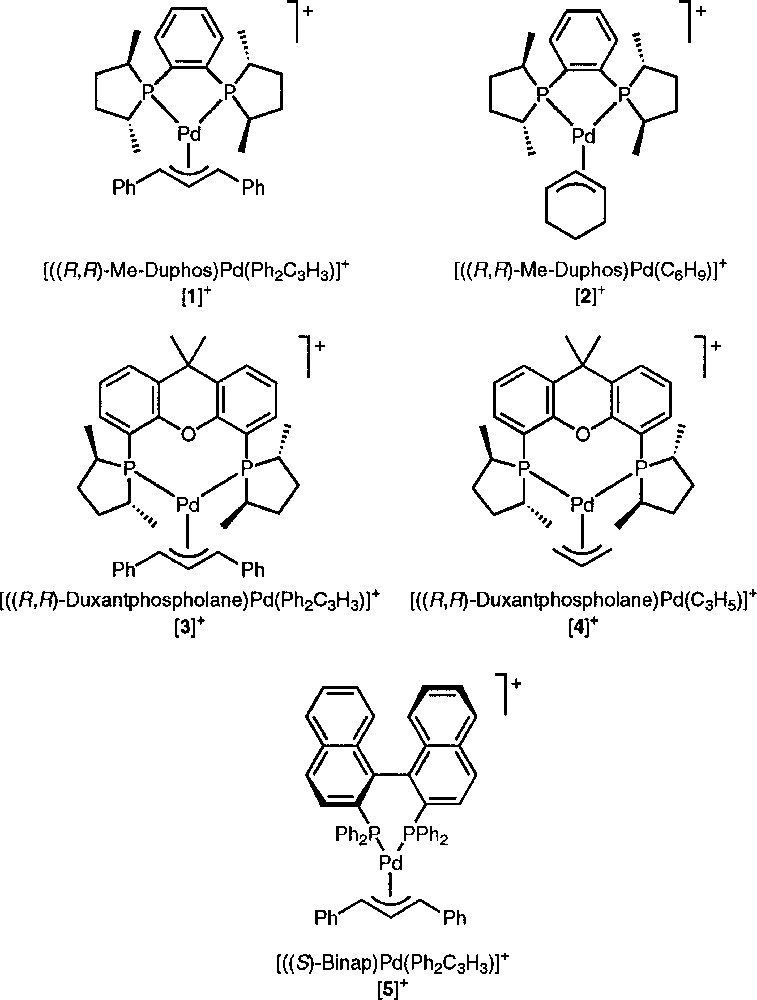
Studied allylic diphosphine palladium (II) complexes.
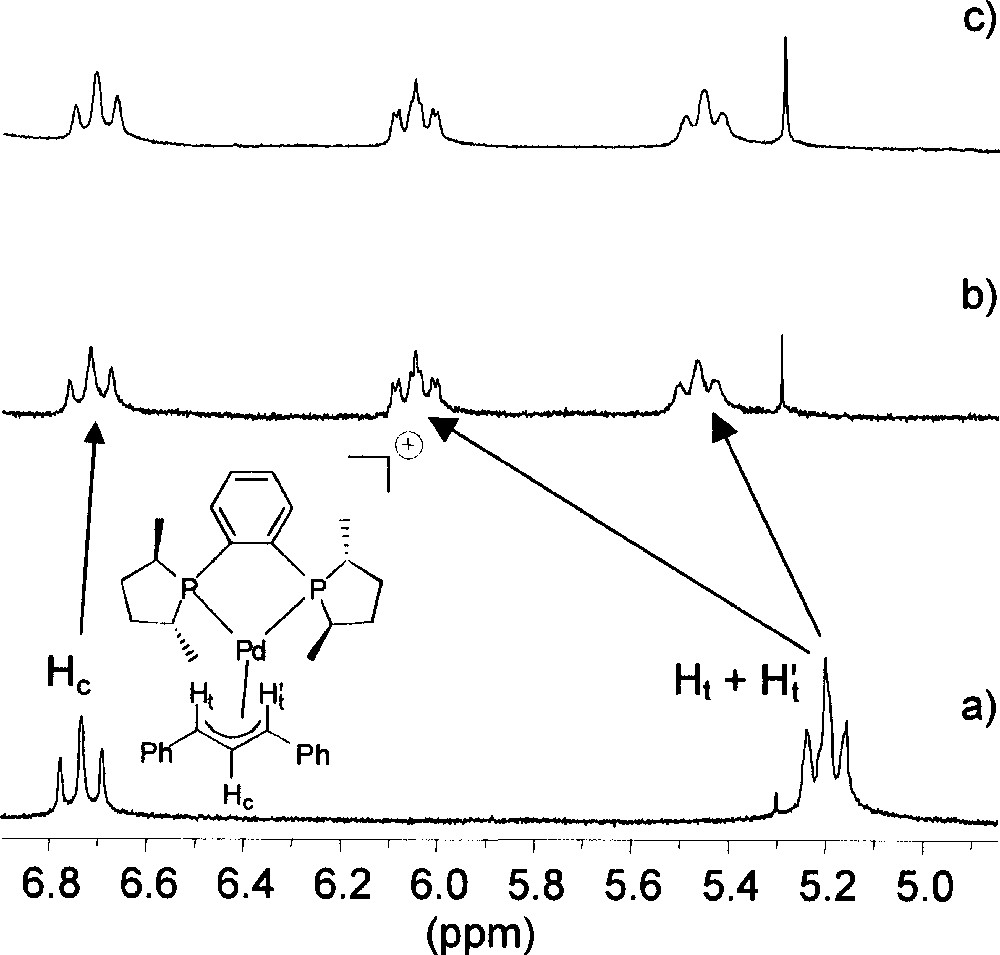
Section of 1H NMR spectra (CDCl3, 298 K, 300 MHz) of (a) complex 〚1〛SbF6; (b) complex 〚1〛SbF6 + 1 equiv PPh4Cl; (c) complex 〚1〛Cl obtained from the reaction of 1 equiv 〚Pd(η3-Ph2C3H3)Cl〛2 with 2 equiv (R,R)-Me-Duphos.
The chemical shifts of some other 1H signals of 〚1〛SbF6 are also modified (although to a lower extent) in the presence of Cl–, without any change of the coupling constants. The values of Δδ for the methyl signals of Me-Duphos are given in Fig. 3. One salient feature of the ‘chloride effect’ is that the nuclei located under the coordination plane are deshielded (+0.12 ≤ Δδ ≤ +0.84 ppm), while those located above are unchanged or little shielded (–0.08 ≤ Δδ ≤ 0 ppm). In parallel, slight modifications of the 31P{1H} NMR spectrum of 〚1〛SbF6 in CDCl3 are observed when 1 equiv Cl– is added. The doublets of the AB system move from 65.5 and 69.0 ppm to 65.9 and 68.3 ppm, and the P–P coupling constant increases from 49 to 53 Hz. We observe that the NMR features of 〚1〛Cl, obtained by reacting 1 equiv 〚Pd(η3-Ph2C3H3)Cl〛2 with 2 equiv (R,R)-Me-Duphos, are exactly identical to those of the addition product of PPh4Cl with 〚1〛SbF6 (see for instance Fig. 2b and c). This suggests that SbF6– effectively behaves as an inert ion, whereas Cl– displays a specific interaction with the cationic palladium complex.
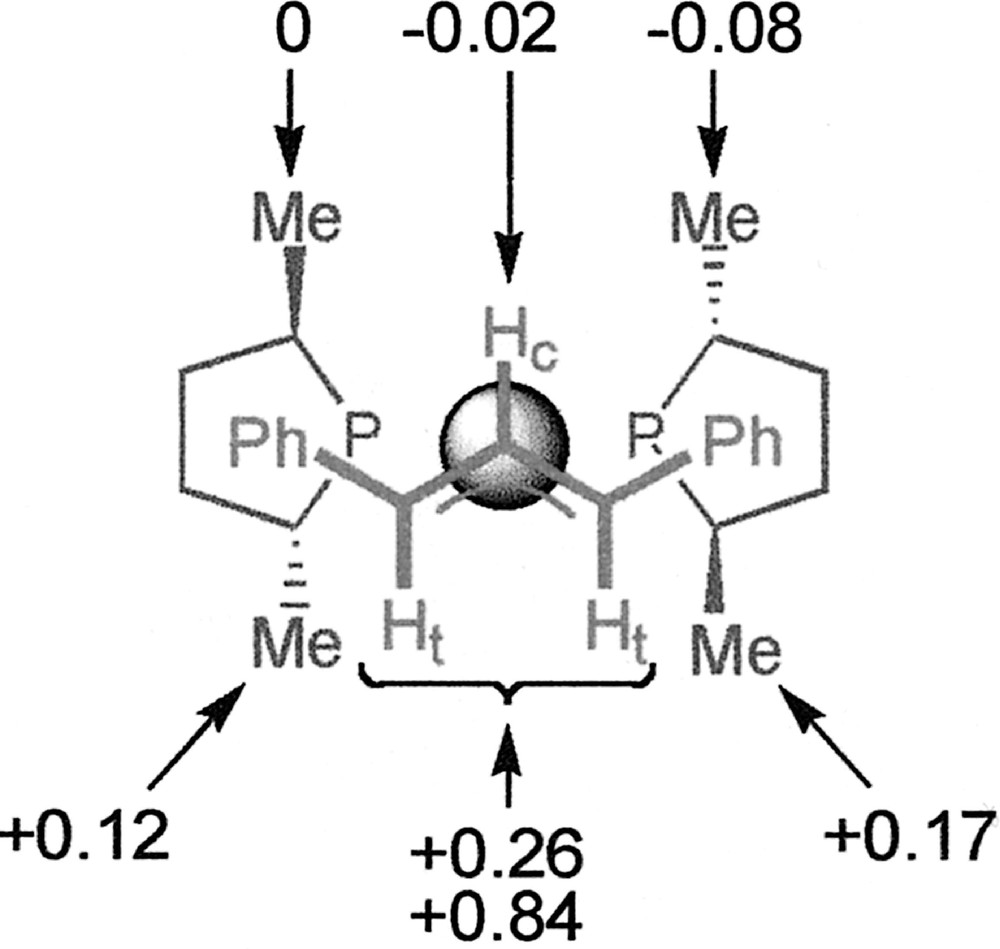
Values of the chemical shift variations (Δδ, ppm) of different proton signals of complex 〚1〛SbF6 upon addition of Cl– in CDCl3 at 298 K. The metallic centre is represented by the sphere.
In other solvents like CD2Cl2 or CD3CN, similar changes on the 1H NMR spectrum of 〚1〛SbF6 are induced by chloride ions, but to a lower extent (Table 1, entries 1–5). The terminal allylic signals move from 0.02 to 0.15 ppm when 1 equiv Cl– is added, whereas the signals of Hc and of the protons of Me-Duphos are unchanged. In CD2Cl2, the Ht signals are shifted back to 5.10 ppm when the temperature is lowered from 298 to 233 K (entries 2–3).
Effect of chloride ions on the 1H NMR allylic signals of palladium allyl complexes.
| Entry | Complex | Solvent | T (K) | equiv PPh4Cl added | Ht | Hc |
| 1 | 〚1〛SbF6 | CD2Cl2 | 298 | 0 | 5.04 (13) | 6.80 (12.9) |
| 5.11 (13) | ||||||
| 2 | 〚1〛SbF6 | CD2Cl2 | 298 | 1 | 5.18 (12) | 6.80 (12.8) |
| 5.26 (13) | ||||||
| 3a | 〚1〛SbF6 | CD2Cl2 | 233 | 1 | 5.10c | 6.77 (12.8) |
| 4 | 〚1〛SbF6 | CD3CN | 298 | 0 | 5.14c | 6.90 (12.9) |
| 5 | 〚1〛Cld | CD3CN | 298 | 0 | 5.16 (10) | 6.91 (12.9) |
| 5.27 (11) | ||||||
| 6 | 〚2〛BF4 | CDCl3 | 298 | 0 | 5.94 | 5.42 (7.2) |
| 6.00 | ||||||
| 7 | 〚2〛BF4 | CDCl3 | 298 | 1 | 5.91 | 5.40 (7.1) |
| 5.99 | ||||||
| 8b | 〚exo-3〛SbF6 | CD2Cl2 | 193 | 0 | 4.68 (10.8) | 6.36 (13.8, 10.3, 2.4) |
| 5.38 (13.7, 9.4) | ||||||
| 9b | 〚exo-3〛SbF6 | CD2Cl2 | 193 | 1 | 4.68 (11.1) | 6.35 (13.7, 10.3, 2.3) |
| 5.38 (13.7, 9.4) | ||||||
| 10b | 〚endo-3〛SbF6 | CD2Cl2 | 193 | 0 | 4.89 (11.1) | 5.18 (12.5) |
| 5.01 (13.0, 9.2) | ||||||
| 11b | 〚endo-3〛SbF6 | CD2Cl2 | 193 | 1 | 4.92 (11.2) | 5.18 (12.5) |
| 5.05 (12.8, 9.7) | ||||||
| 12 | 〚4〛SbF6 | CD2Cl2 | 263 | 0 | 2.44 (11), 2.61 (11)e | 5.17 |
| 4.48, 4.60f | ||||||
| 13b | 〚4〛SbF6 | CD2Cl2 | 263 | 1 | 2.49, 2.67e | 5.18 |
| 4.49, 4.62f | ||||||
| 14 | 〚5〛BF4 | CDCl3 | 298 | 0 | 4.55 (11.5)g | h |
| 15 | 〚5〛BF4 | CDCl3 | 298 | 1 | 4.95 (11.6)g | h |
a Chemical shift data (ppm) of spectra recorded on a 300 MHz spectrometer unless otherwise stated. Numbers in parentheses are coupling constants J (Hz). The signals are multiplets or large signals (no value of J indicated), pseudo-triplets (one value of J), doublets of doublets (two values of J), or doublets of doublets of doublets (three values of J). Ht = terminal allylic protons; Hc = central allylic proton. The data provided for complexes 〚1〛+, 〚3〛+ and 〚5〛+ correspond to overwhelmingly major syn–syn isomers; two isomers exo and endo are observed for 〚3〛+ 〚16–19〛.
b Spectrum recorded at 500 MHz.
c Two superimposed signals.
d Obtained from the reaction of 1 equiv. 〚Pd(η3-Ph2C3H3)Cl〛2 with 2 equiv. (R,R)-Me-Duphos.
e Hanti.
f Hsyn.
g Only one signal visible 〚19〛.
h Not visible.
We tested the effect of Cl– on other palladium allyl complexes, changing either the substituents of the allyl ligand (complex 〚2〛+), the diphosphine ligand (complexes 〚3〛+ and 〚5〛+) or both (complex 〚4〛+) (Table 1, entries 6–15). Incidentally, the spectra of 〚3〛SbF6 were recorded at 193 K in order to visualise the signals of the frozen exo and endo isomers 〚16, 17〛. Likewise, the spectra of 〚4〛SbF6 were recorded at 263 K, so that the classical chloride-induced syn–anti exchange of Pd-allyl complexes 〚13〛 would be frozen. By contrast with complex 〚1〛+, the 1H NMR signals of the other complexes are not much affected by Cl–, the chemical shift variation reaching at the most 0.06 ppm. One exception is complex 〚5〛+, the Ht signal of which is deshielded from 4.55 to 4.95 ppm. In summary, the deshielding effect of chloride ions is essentially observed on 1,3-diphenylallyl complexes.
In close analogy with our results, Godleski et al. have studied the 1H NMR spectrum in CDCl3 of a cationic palladium allyl bis(triphenylphosphine) complex with different counteranions 〚10〛. They found that the allylic proton resonances were about the same with PF6– at room temperature and with Cl– at 208 K, but were comparatively deshielded with Cl– at room temperature. They suggested that the two first cases would correspond to a free ion pair, whereas in the last one Cl– would be tightly bound to the cation (contact ion pair) or coordinated to Pd (pentavalent species). This interpretation may fit as well with our own observations; all the more, the hypothesis of tight ions pairs between Cl– and Pd-allyl complexes has also been formulated by others 〚8, 9, 11, 13〛.
We focused on the analysis of complex 〚1〛Cl, for which the NMR effect was strongest, in order to gather more information concerning its exact nature. Conductivity has proven to be a useful method to study palladium allyl complexes 〚10〛. The value of the molar conductivity of 〚1〛Cl in acetonitrile is 104 Ω–1 cm2 mol–1 at a 2×10–3 M concentration, which falls within the expected range (92–199 Ω–1 cm2 mol–1) for a 1:1 electrolyte in this solvent 〚20〛. By contrast, the complex is non-conducting in chloroform, which is consistent with a close ion pair or a neutral species. We tried to detect a possible Pd–Cl bond by means of FAB–MS analysis or FT–IR spectroscopy. The FAB–MS spectrum of a 1:1 mixture of 〚1〛SbF6 and PPh4Cl in CDCl3 does not exhibit any peak corresponding to a neutral chloride-coordinated species; the highest peak at m/z = 605.3 corresponds to the molecular peak of the 〚1〛+ cation. No Pd–Cl stretching band is either observed on the IR spectrum of 〚1〛Cl in KBr in the 200–400 cm–1 region 〚21〛. Therefore, no evidence of a pentacoordinate complex is provided by those analytical methods. Interestingly, whether Cl– is bound or not to Pd, the Δδ distribution shown in Fig. 3 may suggest that the anion is preferentially localised on one side of the coordination plane.
In general, cationic palladium diphosphine allyl complexes listed in Fig. 1 are far less stable in solution with chloride as anion than with a non-coordinating anion; palladium black deposition and emergence of unknown signals on NMR spectra are observed. For example, complex 〚1〛+ is stable for days in chloroform with SbF6– as counteranion, whereas with Cl– it is reactive (see below).
We endeavoured to isolate the compound 〚1〛Cl in a crystalline form by slow vapour-phase diffusion of pentane at room temperature into a mixture of Me-Duphos and the dimeric complex 〚Pd(η3-Ph2C3H3)Cl〛2 (molar ratio 2:1) dissolved in CH2Cl2. This led to yellow single crystals suitable for an X-ray diffraction study, whereas the supernatant appeared as a cloudy palladium metal suspension. Unexpectedly, their X-ray analysis revealed that the crystallised molecule was the neutral palladium complex 〚((R,R)-Me-Duphos)PdCl2〛 6; one molecule of CH2Cl2 co-crystallises with the complex. The preparation of this complex by another route has been reported very recently 〚22〛. The solid-state structure is depicted in Fig. 4, and the selected bond distances and angles are given in Table 2. The palladium atom is coordinated by the two chlorides and the two phosphorus atoms of Duphos in a classical square-planar geometry. The long Pd–Cl bond distances (2.3706(9) and 2.3790(9) Å) are characteristic of complexes bearing ligands with a strong trans influence 〚23〛, viz. the phospholanes. The weaker trans influence of a chloro ligand compared to an allyl ligand explains that the Pd–P bonds are smaller (2.218 7(9) and 2.220 5(9) Å) than in the related complex 〚(R,R)-Me-Duphos)Pd(η3-Ph2C3H3)〛(CF3SO3) 〚18〛. The other characteristics of the palladacycle (e.g. the P1–Pd–P2 angle, 86.51(3) Å) and of the Duphos ligand are comparable to those of references 〚18, 22〛. The palladium centre and the four donor atoms are almost coplanar, with the palladium lying only 0.016 1(2) Å away from the plane defined by P1, P2, Cl1 and Cl2. This is probably related to the absence of steric interaction between the chloride atoms and the methyl groups borne by Duphos.
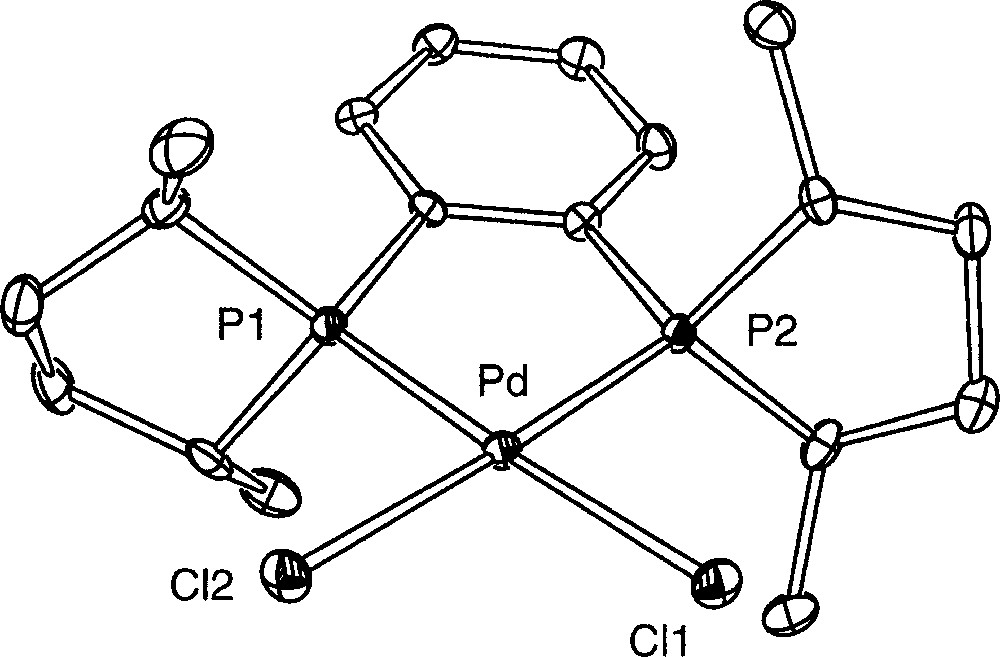
ORTEP drawing of complex 6 showing 30% probability thermal ellipsoids and the atom-numbering scheme; hydrogen atoms and CH2Cl2 are omitted for clarity.
Selected bond distances 〚Å〛 and bond angles 〚°〛 for 6.
| Pd–P1 | 2.2187(9) | Pd–Cl1 | 2.3706(9) |
| Pd–P2 | 2.2205(9) | Pd–Cl2 | 2.3790(9) |
| P1–Pd–P2 | 86.51(3) | Cl1–Pd–Cl2 | 95.46(3) |
| P1–Pd–Cl2 | 89.37(3) | P1–Pd–Cl1 | 175.06(3) |
| P2–Pd–Cl1 | 88.64(3) | P2–Pd–Cl2 | 175.85(3) |
The crystalline complex 6 was isolated in 46% yield and completely characterised. Its NMR features confirm the presence of a C2 symmetry axis, with two methyle and two methyne signals on the 1H and 13C NMR spectra, and one singlet at 95.9 ppm on the 31P{1H} NMR spectrum. On the 13C NMR spectrum, the methyne carbons appear as two second-order signals at 39.1 and 43.8 ppm. From the interpretation proposed for similar signals in complex 〚(R,R)-Me-Duphos)Pd(η3-Ph2C3H3)〛(CF3SO3) 〚18〛, we can estimate the values of |1JCP + 3JCP| as 30 and 31 Hz.
In order to understand what was the fate of the allyl ligand, we checked the spontaneous evolution of 〚1〛Cl in different solvents at room temperature. For that purpose, 2 equiv Me-Duphos and 1 equiv 〚Pd(η3-Ph2C3H3)Cl〛2 were mixed in CDCl3 or CD3CN and the solution periodically analysed by 1H and 31P NMR. The evolution was the same in both solvents, but at a different rate, as the reaction was complete within 2 days in CDCl3 and 10 days in CD3CN. With time, palladium metal deposited in the tube and the 1H and 31P signals of 6 appeared, whereas those of 〚1〛Cl decreased in intensity. In the final mixture, the allyl ligand has been converted into chalcone (89%), the coupling product 1,3,4,6-tetraphenylhexa-1,5-diene (9%) and diphenylpropene (2%) (Fig. 5). Thus the largest part of the allyl ligand has been oxidised, probably by some dioxygen present in solution. The trans/cis ratio of chalcone depends on the solvent (CDCl3, 67:33; CD3CN, 57:43); we note that the proportion of the thermodynamically less stable cis isomer is unexpectedly high. It is surprising that the allylic moiety could be oxidised in such soft conditions, insofar as the oxidation of a palladium allyl complex to a ketone generally requires a strong oxidant 〚24〛. In connection with our observations, examples of palladium-catalysed coupling of allylic acetates 〚25, 26〛 or carbonates 〚27〛, or of coupling of allyl ligands from palladium complexes 〚28, 29〛, have been reported.

Spontaneous transformation of complex 〚1〛Cl at RT in CDCl3 or CD3CN.
Another kind of transformation of 〚1〛Cl was observed when the crystallisation took place in acetonitrile at low temperature. Crystals suitable for an X-ray analysis were obtained from the slow cooling to 250 K of a solution of 〚1〛Cl in CD3CN. At this temperature, the crystallised species was the ion pair 〚1〛〚PdCl2(η3-Ph2C3H3)〛 (Figs. 6 and 7, Table 3). A similar transformation upon attempted precipitation of a cationic palladium allyl complex with Cl– as counteranion has been reported by Crociani et al. 〚11〛. Separate ((R,R)-Me-Duphos)(η3-diphenylallyl)palladium (II) cations and dichloro(η3-diphenylallyl)palladium (II) anions are present in the unit cell. The cation exhibits no significant structural difference with 〚1〛CF3SO3 〚18〛. The bond distances and angles of the anionic complex are globally similar to those of the reported X-ray structures of 〚PdCl2(η3-C3H5)〛– 〚9, 23〛, except the Cl1–Pd2–Cl2 angle, which is slightly larger (99.90(7)° instead of 95.8(1)° or 97.9(2)°). In a similar crystallographic structure reported by Tresoldi et al. 〚9〛, one chloro ligand of the anion weakly coordinates the palladium of the cation with a 3.07 Å distance. It is not the case here, the closest chloro (Cl1) lying 3.517 Å away from Pd1.
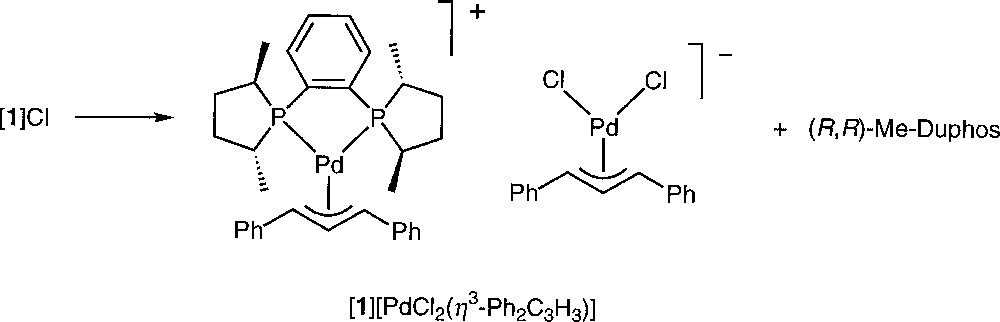
Spontaneous transformation of complex 〚1〛Cl at 250 K in CD3CN.
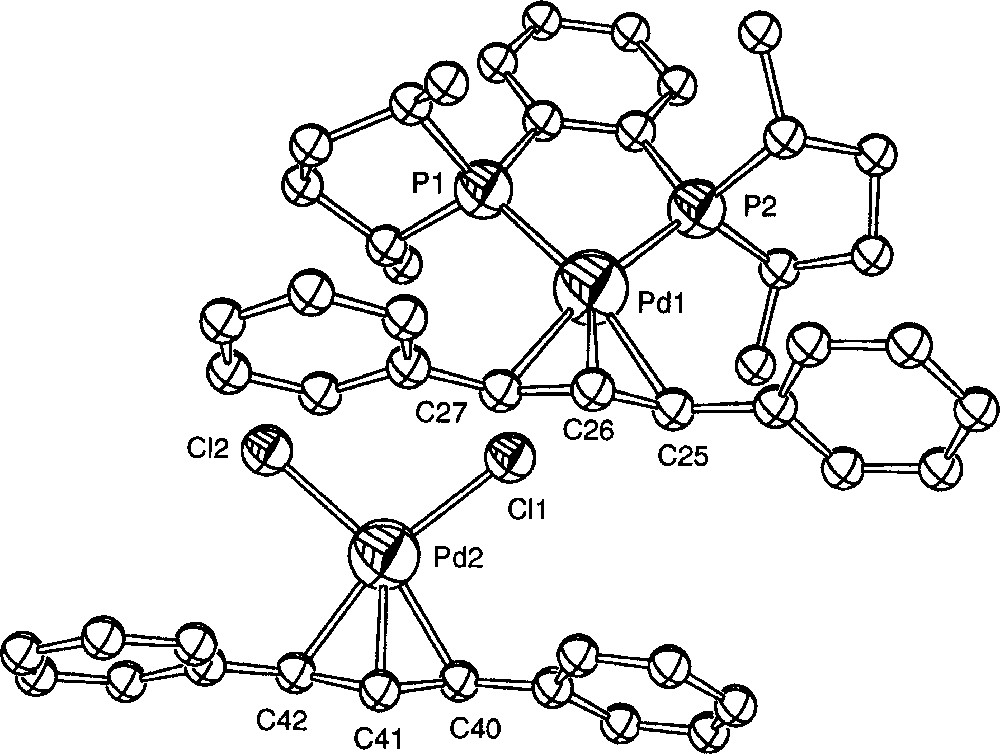
ORTEP drawing of compound 〚1〛〚PdCl2(η3-Ph2C3H3)〛 showing 30% probability thermal ellipsoids and the atom-numbering scheme; hydrogen atoms are omitted for clarity.
Selected bond distances 〚Å〛 and bond angles 〚°〛 for 〚1〛〚PdCl2(η3-Ph2C3H3)〛.
| Pd1–P1 | 2.292(2) | Pd2–Cl1 | 2.380(2) |
| Pd1–P2 | 2.270(2) | Pd2–Cl2 | 2.357(2) |
| Pd1–C25 | 2.234(6) | Pd2–C40 | 2.156(7) |
| Pd1–C26 | 2.211(6) | Pd2–C41 | 2.109(7) |
| Pd1–C27 | 2.245(7) | Pd2–C42 | 2.161(8) |
| C25–C26 | 1.402(9) | C40–C41 | 1.42(1) |
| C26–C27 | 1.37(1) | C41–C42 | 1.41(1) |
| P1–Pd1–P2 | 86.51(6) | Cl1–Pd2–Cl2 | 99.90(7) |
| P1–Pd1–C27 | 103.0(2) | Cl1–Pd2–C40 | 96.8(2) |
| P2–Pd1–C25 | 104.6(2) | Cl2–Pd2–C42 | 95.2(2) |
| C25–Pd1–C27 | 65.5(3) | C40–Pd2–C42 | 68.2(3) |
| C25–C26–C27 | 121.9(6) | C40–C41–C42 | 117.8(7) |
3 Conclusion
The first conclusion we can draw from our study is that the structure of the coordination sphere of diphosphine Pd allyl complexes is not strongly altered by chloride ions. However, and in accordance with previously reported observations on other Pd allyl complexes 〚10〛, the protons of the complexes are sometimes deshielded. Yet in our case this deshielding effect of Cl– is not systematic: for instance, with Me-Duphos as the diphosphine, it is strong on the 1,3-diphenylallyl complex 〚1〛+, but non-existent on the cyclohexenyl complex 〚2〛 +. From a thorough analysis of compound 〚1〛Cl, we conclude that this compound is present as separate ions in acetonitrile, whereas it is under the form of a tight ion pair or a neutral pentavalent complex in chloroform.
The presence of chloride ions makes the Pd allyl complexes reactive in solution; this instability may be connected with the fact that Cl– can improve the performance of allylic alkylation Pd catalysts 〚2, 17〛. Investigations on the nature of the transformation products at room temperature in the case of compound 〚1〛Cl reveal that Cl– pushes the allyl ligand away from the coordination sphere. This leads to the formation of palladium black and of complex 6, whereas most of the diphenylallyl ligand is oxidised into chalcone. On the reverse, crystals of 〚1〛〚PdCl2(η3-Ph2C3H3)〛 form upon cooling a solution of 〚1〛Cl in acetonitrile.
4 Experimental
The NMR spectra were obtained at room temperature (unless otherwise indicated) on Bruker spectrometers. 1H NMR spectra were recorded at 300.16 MHz (AC-300 instrument) or 500.13 MHz (ARX-500) and referenced to SiMe4. 13C{1H} NMR spectra (broadband decoupled) were recorded at 50.32 MHz (AC-200) and referenced to SiMe4; the assignments were supported by DEPT-135°. 31P{1H} NMR spectra (broadband decoupled) were recorded at 121.51 MHz (AC-300) and referenced to 85% aqueous H3PO4. The NMR tubes were prepared at a concentration ca 15–25 mM under a nitrogen atmosphere using a Vacuum Atmospheres glovebox equipped with Dri-Train HE-493 inert gas purifier; the solutions were stored in the air after the recording of the spectra. The FT–IR spectra were recorded on a Perkin-Elmer 1600 Series spectrometer on KBr pellets. The FAB–MS spectra (NBA matrix) and elemental analyses were carried out by the corresponding facilities at the ‘Fédération de recherche de chimie, université Louis-Pasteur’, Strasbourg, France. The ligands (R,R)-Me-Duphos and (S)-Binap were purchased from Strem and used as received. The complex 〚Pd(η3-Ph2C3H3)Cl〛2 was prepared following a reported procedure 〚30〛. The complexes 〚1〛SbF6, 〚2〛BF4, 〚3〛SbF6, 〚4〛SbF6 and 〚5〛BF4 were prepared following reported procedures 〚16–19〛. The 1H NMR signals of 〚1〛SbF6 have been assigned from NOE effects observed on a 1H–1H ROESY spectrum.
4.1 Preparation and transformations of 〚1〛Cl at room temperature
To a suspension of 10 mg (0.015 mmol) 〚Pd(η3-Ph2C3H3)Cl〛2 in 1 ml CDCl3 (or CD3CN) was added a solution of 10 mg (0.032 mmol) (R,R)-Me-Duphos in 1 ml CDCl3 (CD3CN). The mixture was stirred for 10 min at room temperature until a clear yellow solution was obtained. After several days, the different products were identified by 1H and 31P NMR, by comparison with an authentic sample (cis- and trans-chalcone), with literature data (1,3,4,6-tetraphenylhexa-1,5-diene, 1,3-diphenylpropene) 〚31〛 or with NMR data given in § 4.2 (6).
4.2 Complex 〚((R,R)-Me-Duphos)PdCl2〛 (6)
A solution of 〚1〛Cl was prepared as described above using CH2Cl2 as solvent. The volume was reduced under reduced pressure to 0.5 ml, and pentane was slowly added to the solution. Yellow crystals formed after one week, together with a palladium black suspension. The crystals were washed with diethyl ether and dried under vacuum (7 mg, 0.014 mmol, 46%).
1H NMR (CD3CN, 300 MHz, 298 K): δ (ppm) 0.90 (dd, 6H, 3JHP = 16.8 Hz, 3JHH = 7.3 Hz, CH3); 1.52 (dd, 6H, 3JHP = 19.5 Hz, 3JHH = 6.8 Hz, CH3); 1.63–2.50 (m, 8H, CH2); 2.77 (m, 2H, CH); 3.49 (m, 2H, CH); 7.65–7.90 (m, 4H, Har).
13C{1H} NMR (CDCl3, 50 MHz, 298 K): δ (ppm) 14.8 (s, CH3); 17.9 (virtual t, |2JCP + 4JCP| = 6 Hz, CH3); 36.6 (s, CH2); 37.9 (s, CH2); 39.1 and 43.8 (second-order signals, CH); 133.3 (virtual t, |2JCP + 3JCP| = 20 Hz, CHar); 133.6 (s, CHar); 142.1 (virtual t, |1JCP + 2JCP| = 74 Hz, Car).
31P{1H} NMR (CD3CN, 121.5 MHz, 298 K) : δ (ppm) 95.9 (s, 2P).
Elemental analysis calcd for C18H28Cl2P2Pd: C, 44.70; H, 5.84; P, 12.81; found C, 44.61; H, 6.02; P, 13.06.
4.3 Reactivity of 〚1〛Cl at low temperature
A solution of 〚1〛Cl in CD3CN was prepared as described above and slowly cooled to 250 K. After 2 days, yellow crystals suitable for X-ray analysis were obtained.
4.4 X-ray crystallography
Details of data collection parameters and refinements results are listed in Table 4. The structures were solved using direct methods. After refinement of the non-hydrogen atoms, difference-Fourier maps revealed maximums of residual electron density close to positions expected for hydrogen atoms. Hydrogen atoms were introduced as fixed contributors at calculated positions (C–H = 0.95 Å, B(H) = 1.3 Beqv). Final difference maps revealed no significant maximums. All calculations were done using the Nonius OpenMoleN package 〚32〛. Neutral atom scattering factor coefficients and anomalous dispersion coefficients were taken from a standard source 〚33〛.
Crystal and refinement data for complexes 6 and 〚1〛〚PdCl2(η3-Ph2C3H3)〛.
| Compound | 6·CH2Cl2 | 〚1〛〚PdCl2(η3-Ph2C3H3)〛 |
| Formula | C18H28Cl2P2Pd·CH2Cl2 | C15H13Cl2Pd·C33H41P2Pd |
| Molecular weight | 568.61 | 976.62 |
| Color | Yellow | Yellow |
| Crystalline system | Orthorhombic | Monoclinic |
| a (Å) | 9.7387(3) | 31.979(1) |
| b (Å) | 13.4181(3) | 12.2296(3) |
| c (Å) | 18.0813(5) | 11.6346(4) |
| β (deg) | 107.668(5) | |
| V (Å3) | 2362.8(2) | 4335.6(5) |
| Z | 4 | 4 |
| Dcalc (g cm–3) | 1.60 | 1.50 |
| F000 | 1152 | 1992 |
| Wavelength (Å) | 0.71073 | 0.71073 |
| μ (mm–1) | 1.376 | 1.059 |
| Space group | P212121 | C121 |
| Diffractometer | KappaCCD | KappaCCD |
| Crystalline dimensions (mm) | 0.20 × 0.18 × 0.16 | 0.20 × 0.16 × 0.12 |
| Temperature (K) | 173 | 173 |
| Radiation | Mo Kα graphite monochromated | Mo Kα graphite monochromated |
| Scan mode | ‘phi scans’ | ‘phi scans’ |
| hkl limits | –12,12/–17,17/–23,23 | –41,41/–15,12/–15,15 |
| θ limits (°) | 2.5/27.48 | 2.5/27.48 |
| No. of data measurements | 5417 | 8879 |
| No. of data with I > 3 σ(I) | 2650 | 4506 |
| No. of variables | 235 | 486 |
| R | 0.025 | 0.031 |
| Rw | 0.028 | 0.048 |
| Largest peak in final difference (e Å–3) | 0.699 | 1.243 |
| GOF | 1.081 | 1.017 |
Supplementary material
The supplementary material has been sent to the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK 〚Fax: + 44-1223/336-033; E-mail: deposit@ccdc.cam.ac.uk〛 as supplementary material CCDC 177889 / CCDC 177943, and can be obtained by contacting the CCDC.
Acknowledgements
We thank Shailesh Ramdeehul for helpful discussions at the beginning of this work. Financial support from the French ‘Centre national de la recherche scientifique’ is gratefully acknowledged. GM is indebted to the French ‘Ministère de l’Éducation nationale, de l’Enseignement supérieur et de la Technologie’ for a grant.