

Ligand optimisation in asymmetric transition metal catalysis often entails the necessity to synthesise derivatives of a given structural lead motif 〚1–5〛. Another way to tune ligands is to incorporate two sources of chirality, giving rise to two diastereomers that can then be checked for matched or mismatched interaction in a given catalytic asymmetric transformation 〚1–7〛. In the case of modular ligands composed of three or more stereocentres, four and more diastereomers are possible, as in systems comprising several amino acids 〚8–10〛. Recently we reported the first case of chiral phosphite/phosphonite ligands having two centres of chirality 〚11〛. We now report the synthesis and application of modular phosphite/phosphonite ligands in which three elements of chirality give rise to four possible diasteromers. Significant differences in catalyst activity and enantioselectivity of the corresponding ligand/metal complexes result, depending upon the particular diastereomer used.
The starting point of our synthesis is the commercially available bromide (S)-1, which underwent smooth bis-lithiation and phosphorylation with formation of (S)-3. In situ phosphorylation with (S)-4 afforded (S,S)-5, which was reacted in crude form with (R)-BINOL. In contrast to the previous reactions that proceeded nearly quantitatively, the reaction with BINOL turned out to be somewhat inefficient, even in the presence of tetrazole as an additive 〚12〛. Nevertheless, pure (S,S,R)-6 was obtained in 36% overall yield following recrystallisation 〚13〛 (Fig. 1).
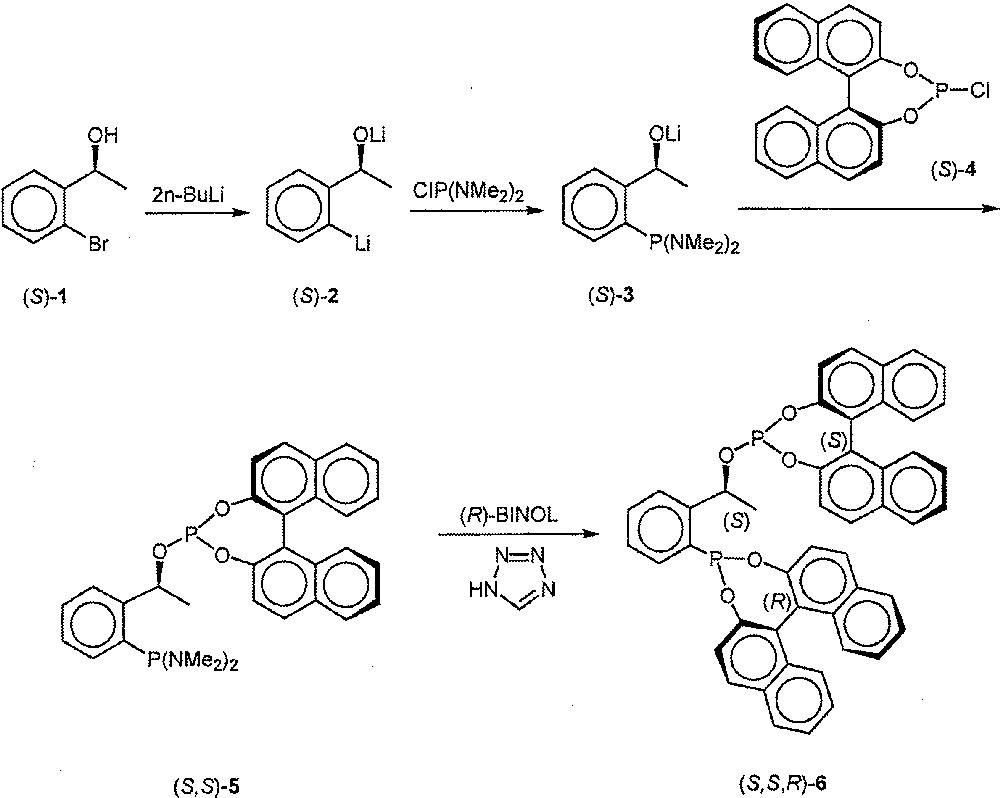
Synthesis of (S,S,R)-6 from (S)-1.
Synthesis of (S,S,R)-6 and analytical data. Under an Ar-atmosphere the mixture of (S)-1-(2-bromophenyl)ethanol (1) (936 mg; 4.66 mmol) in 15 ml of diethyl ether was cooled to -50°C and treated with n-butyllithium (9.3 mmol). The mixture was stirred for 15 min and then allowed to reach room temperature (30 min). After 4 h it was cooled to –80°C and treated dropwise with bis(dimethylamino)chlorophosphine (720 mg; 4.66 mmol). Then the mixture was warmed to room temperature and stirred overnight. The solvent was stripped off, THF (25 ml) added, and the yellow solution added dropwise to a cooled (–80°C) solution of (S)-4 (1.63 g; 4.66 mmol) in 20 ml of THF. After stirring at room temperature overnight, the solvent was stripped off and diethyl ether added (30 ml). Solids were allowed to precipitate and the solution was filtered, the process being repeated four times. The solvent of the combined solutions was stripped off and the foamy residue dried in vacuo. It was dissolved in CH2Cl2 (3 ml) and CH3CN (30 ml) and treated with (R)-BINOL (1.33 g; 4.65 mmol) in CH3CN (4 ml). The solution was cooled to 4°C, treated with tetrazole in CH3CN (2.33 mmol, 50%) and stirred for 2 h at room temperature. The solution was removed and the residue washed twice with CH3CN (8 ml) and dried in vacuo. The colourless solid was recrystallised from CH2Cl2/CH3CN, yielding an analytically pure amorphous powder (1.26 g; 36%). 31P NMR (CD2Cl2): δ (ppm) = 176.0 (d, Jpp = 7.0 Hz, 144.0 (d, Jpp = 7.0 Hz). HRMS (EI): found 750.172 116; calculated 750.172 501.
Analytical data of the other diastereomeric ligands. (S,S,S)-6: 31P NMR (CD2Cl2): δ (ppm) = 180.5 (d, JPP = 4.4 Hz, 152.3 (d, JPP = 4.4 Hz); elemental analysis: found C 77.32, H 4.20, P 8.09; calculated C 76.79, H 4.29, P 8.25. (R,S,S)-6: 31P NMR (CD2Cl2): δ (ppm) = 176.6 (d, JPP = 3.6 Hz), 139.0 (d, JPP = 3.6 Hz); elemental analysis: found C 74.85, H 4.66, P 8.55; calculated 76.79, H 4.29, P 8.25. (R,S,R)-6: 31P NMR (CD2Cl2): δ (ppm) = 176.1 (d, JPP = 8.9 Hz), 144.3 (d, JPP = 8.9 Hz); elemental analysis: found C 75.90, H 4.51, P 8.48; calculated C 76.79; H 4.29, P 8.25 〚13〛.
Since the absolute configuration of the building blocks 4 and BINOL can be chosen as desired, the synthesis of the three other diastereomers proceeded analogously (Fig. 2).
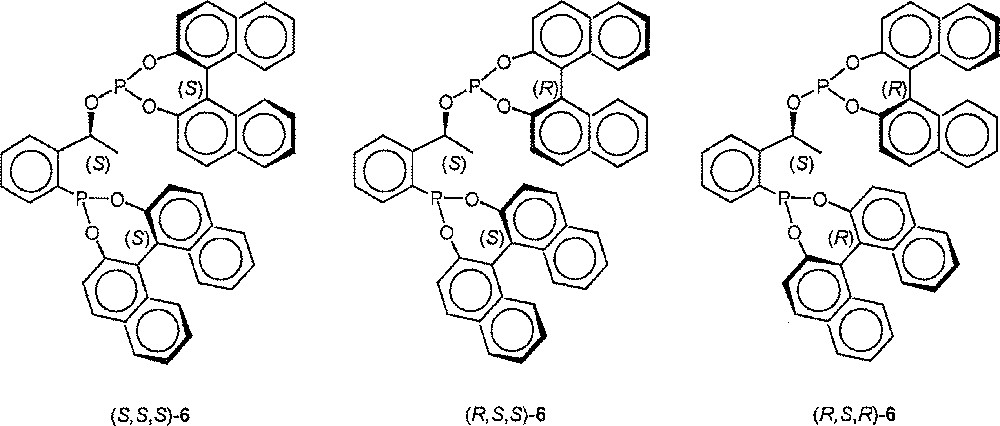
Structures of (S,S,S)-6, (R,S,S)-6, (R,S,R)-6.
We first tested the four ligands in the Cu(OTf)2-catalysed conjugate addition of Et2Zn to cyclohexenone 7 〚14–29〛 (Fig. 3). Large differences in catalyst activity and enantioselectivity were observed. With (R,S,S)-6: conversion = 69%, ee = 16% (R)-product 8; with (S,S,S)-6: conversion = 100%, ee = 77% (S); with (S,S,R)-6: conversion = 96%, ee = 15% (S); with (R,S,R)-6: conversion = 28%; ee = 15% (R). Thus, all three elements of chirality are crucial in determining overall catalyst performance, (S,S,S)-6 representing the best combination.
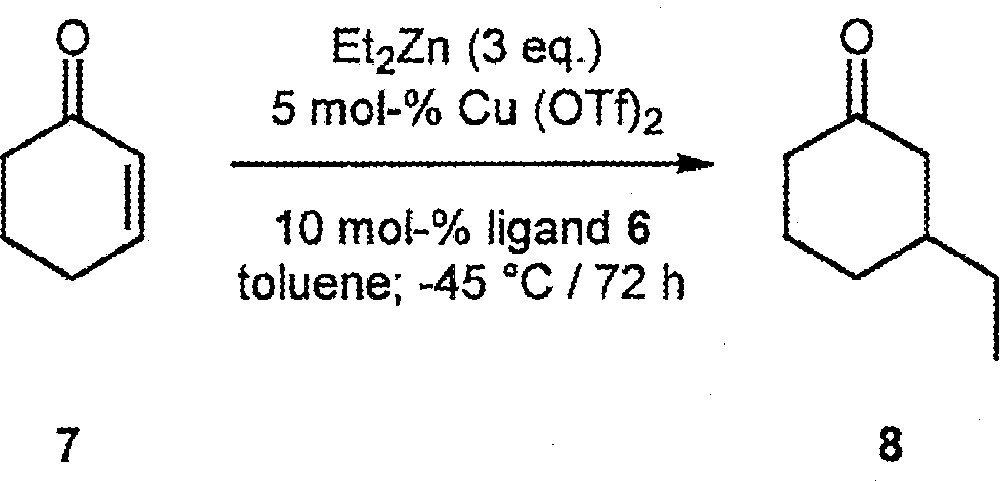
Cu(OTf)2-Catalysed conjugate addition of Et2Zn to cyclohexenone 7.
We next used the four ligands in the Pd-catalysed allylic substitution 9 → 11 〚30, 31〛 (Fig. 4). Again, significant differences in activity and enantioselectivity were observed as a function of the ligand, the best one in this case being diastereomer (S,S,R)-6: conversion = 100%, ee = 72% (S), the others showing poor performance, with (R,S,S)-6: conversion = 2%, ee = 28% (R); with (R,S,R)-6: conversion = 100%, ee = 0%; with (S,S,S)-6: conversion = 100%; ee = 11% (R).

Pd-Catalysed allylic substitution 9 → 11.
Finally, the Rh-catalysed asymmetric hydrogenation of itaconic acid dimethyl ester 12 was carried out 〚1–5〛. In this case, (S,S,R)-6 turned out to be the optimal ligand; conversion = 100%, ee = 79% (S); with (R,S,S)-6: conversion = 100%, ee = 54% (R); with (R,S,R)-6: conversion = 74%; ee = 54% (S); with (S,S,S)-6: conversion = 100%; ee = 28% (R) (Fig. 5).
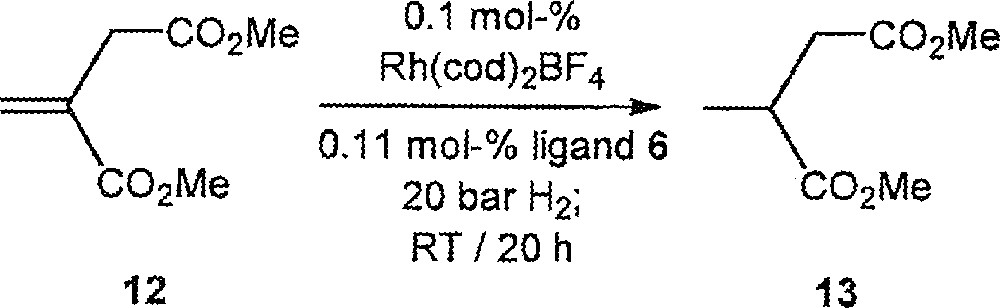
Rh-Catalysed asymmetric hydrogenation of itaconic acid dimethyl ester 12.
In summary, the particular ligands described herein are less efficient than other known systems. However, the principle that ligand tuning is possible by preparing and testing all four diastereomers of a given ligand containing three sources of chirality has been amply illustrated using a phosphite/phosphonite system. Of particular significance is the fact that large differences in catalyst activity are observed in a series of four diastereomeric ligands, in addition to pronounced differences in enantioselectivity. These observations are of considerable theoretical interest. On the practical side, it is likely that more efficient ligands of this general type can be prepared, offering the possibility to match a given substrate with the optimal diastereomeric ligand 〚1–5, 32〛.