

 CC-BY 4.0
CC-BY 4.0
Chiral pure phosphotriesters (phosphates) are of great rarity in catalysis, compared to phosphanes [1], phosphites [2], phosphoric acids or phosphate metal salts [3, 4, 5], and phosphoramidites [6]. Indeed, to date only two examples of their use as organocatalysts are known, which exploit the axial chirality of the BINOL backbone [7, 8]. Ishihara’s group has reported the N-iodolactonization of 4-arylmethyl-4-pentenoic acids catalyzed by BINOL-derived triaryl phosphates as nucleophilic catalysts [7]. Luo and coworkers have applied BINOL-derived trityl phosphates in carbocation catalysis, effective for Friedel–Crafts, inverse electron-demanding hetero-Diels–Alder, and carbonyl-ene reactions [8].
Previously reported chiral phosphotriesters A–F.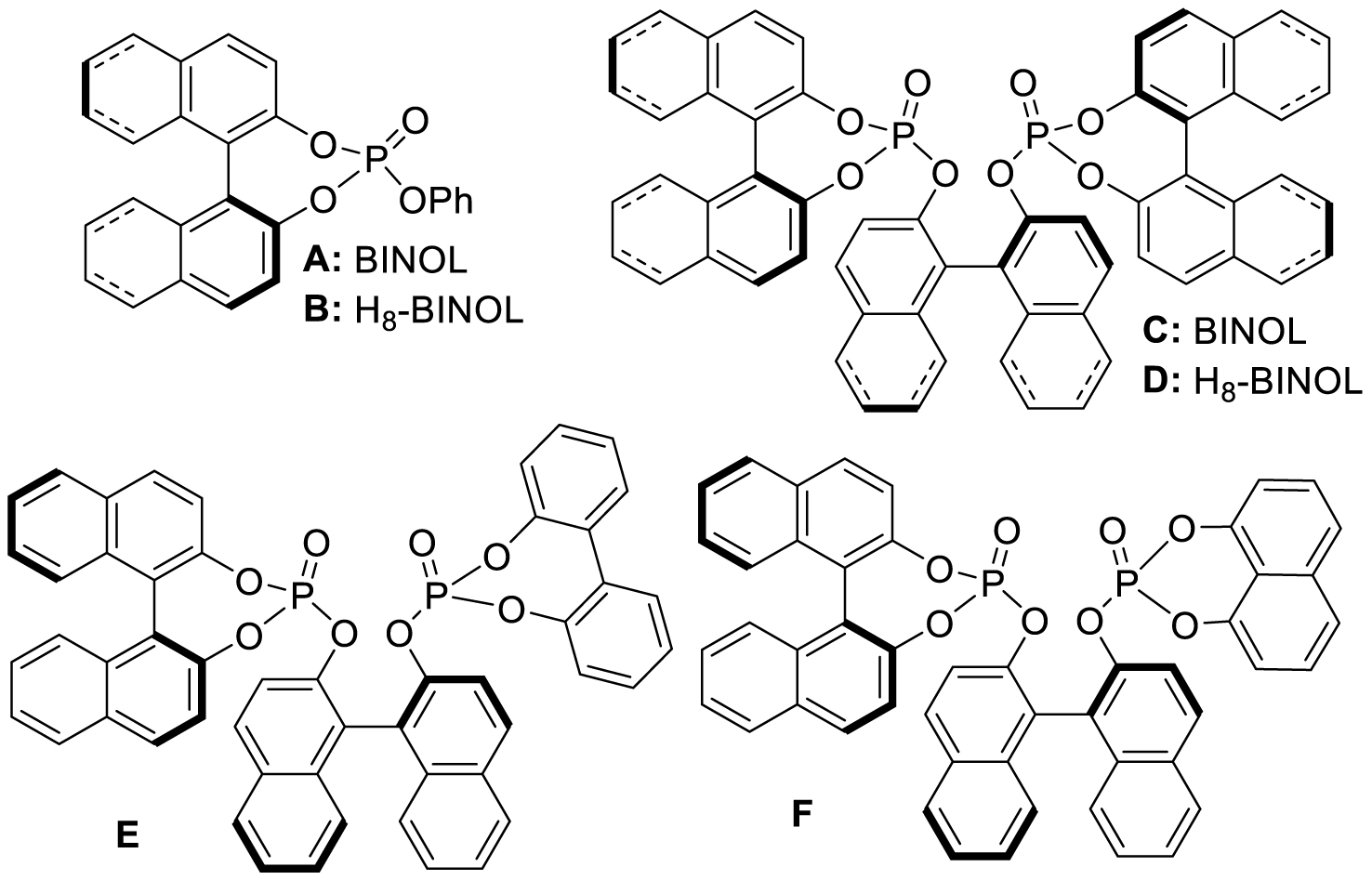
When we started to be interested in pure phosphotriesters as ligands for transition metal catalysis, their use for this purpose was almost unknown and limited to only one example, namely the Pd/phosphate-catalyzed oxidative coupling between arylboronic acids and alkynes, reported by Miura and coworkers [9]. We have recently contributed to this field by disclosing the versatile role of triphenylphosphate in two racemic processes: the Ti-promoted diethylzinc addition to aldehydes and the Zn-catalyzed hydrosilylation of ketones [10]. For thes latter, in a seminal contribution in 1999, Mimoun’s group has disclosed for the first time the use of Zn/chiral secondary diamine complexes for the reduction of ketones in the presence of hydrosilanes, which enable them to reach 80% ee [11]. Since then, other systems still based on N-containing ligands have been reported [12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31], incorporating either diversified diamines or diimines, N,S- or N,O-chelating ligands, or Pybox, and excellent conversions and enantiomeric excesses on a wide range of substrates were thus achieved.
We have chosen to address our efforts toward the challenge of introducing chiral phosphotriesters as ligands in the Zn-catalyzed enantioselective hydrosilylation of prochiral ketones. To the best of our knowledge, this is the first example of their application in the enantioselective version of this transformation, and more widely no examples are provided on the use of chiral phosphotriesters as ligands in transition metal catalysis.
To achieve this, the BINOL-based phosphates A–F (Scheme 1), which have been published elsewhere by us [32], were employed, together with the newly prepared compounds G–I (Scheme 2).
The reaction of 3-I-(R)-BINOL [33] (1) with commercially available phenyl phosphorodichloridate (2) in the presence of Et3N smoothly led to the formation of a 50:50 mixture of two diastereomers G and H in 90% yield, separable by column chromatography on silica gel. Single-crystal X-ray analyses confirmed their structures and unambiguously established the central chirality at the P-atom to be (Sp) and (Rp), for G and H, respectively (Figure 1)1. Following a different synthetic pathway [32], pre-mixing in basic conditions, PCl3 with (S)-BINOL (3), delivered the corresponding phosphorochloridite, which in turn reacted with ethylene glycol to give the desired bisphosphite. The latter underwent in situ oxidation under mild conditions with a 5% NaOCl aqueous solution to afford the corresponding phosphotriester I in 58% overall yield.
Synthesis of phosphates G, H, and I.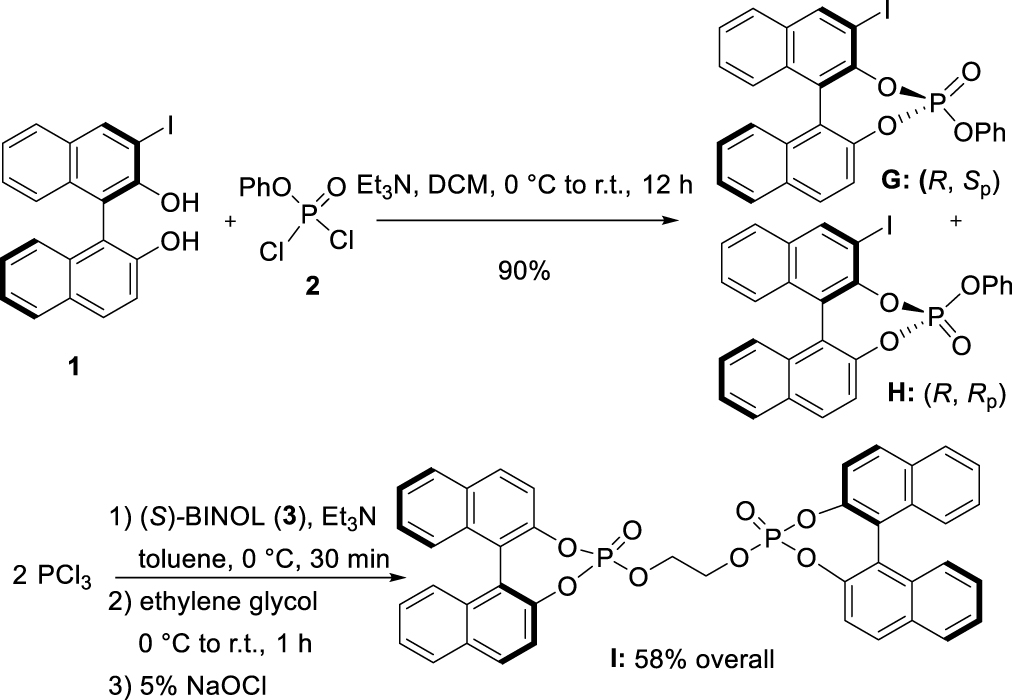
With these chiral ligands in hand, we turned our attention toward their application in the Zn-catalyzed hydrosilylation of acetophenone (4a), chosen as the model substrate (Table 1). Using the reaction conditions optimized with non-chiral phosphates [10], namely 1.5 equiv. of diethoxymethylsilane (DEMS), 5 mol% of ZnEt2, and 5 mol% of ligand in THF, a rapid screening of ligands A–I was conducted. It has to be underlined that the use of any other hydrosilylating agent, in particular PMHS, was detrimental for the reactivity leading to the desired 1-phenylethanol (5a) in only 69% conversion. When the monophosphates A and B derived from (S)-BINOL and (S)-H8-BINOL, respectively, were used as donors for diethylzinc, 4a was reduced to 5a almost without stereochemical control (Table 1, entries 1 and 2). When ligands G and H possessing both axial and central chiralities were used, the reactivity was maintained, since 4a was fully converted into 5a in 79% and 84% yield, respectively (Table 1, entries 3 and 4). Moreover, a kind of matched/mismatched effect was observed regarding the enantioselectivity: 5a was obtained in 18% ee with ligand G of absolute configuration (R, Sp), whereas a racemate was formed with its diastereomer H of absolute configuration (R, Rp). The use of bisphosphates changed the scenario; when BINOPHAT C was employed, 4a was completely consumed and 5a was recovered in 92% yield and 34% ee (Table 1, entry 5). We then tried to decrease the catalytic amount of ligand C to 2.5 mol%, but unfortunately, even if the conversion was still high, the enantiomeric excess dropped to 27% (Table 1, entry 6). Comparison of our set of data for this compound with those reported in the literature [13], and with chiral HPLC analysis of commercially available enantiopure (S) and (R) 1-phenylethanol, confirmed the (S) stereochemistry at the carbon stereocenter. We tried to better control the enantioselectivity by using the symmetric H8-BINOPHAT D (Table 1, entry 7), but Unfortunately, in this case no reaction occurred. This is probably due to the difference in the dihedral angle values of the chiral axis between the aromatic and the partially hydrogenated ligands, preventing the Zn-center to approach the P=O coordination sites of D. Therefore, other structures containing BINOL such as the unsymmetrical non-homo-BINOL ligands E and F were tested. For both, the reactivity (5a was isolated in 88% and 80% yield, respectively) as well as the enantioselectivity was recovered, although thes latter was decreased when compared with C (18% and 16% ee, respectively) as shown in entries 8 and 9 of Table 1. Finally, with bisphosphate I the desired product was obtained in 69% yield but in racemic form (Table 1, entry 10). This can be attributed to the flexibility of the ethyl linker, which precludes the bisphosphotriester to act as bidentate ligand, giving therefore comparable results to those of monophosphates.
ORTEP representations of compounds G (left) and H (right) at 50% thermal ellipsoids.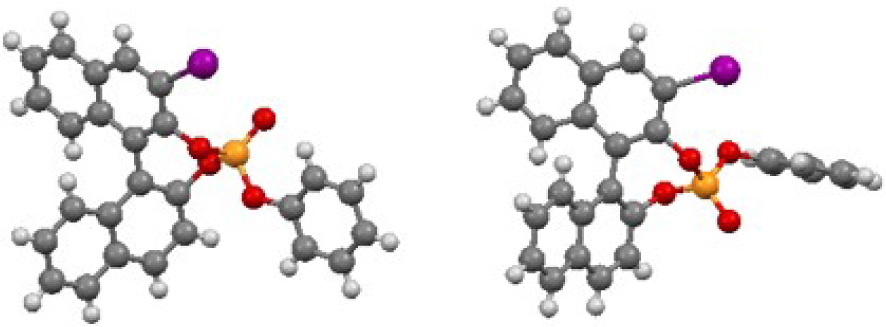
Study for the asymmetric hydrosilylation of ketonesa
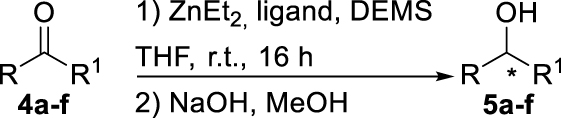
| Entry | Product | Ligand | Conv. (%)b | e.e. (%)c |
|---|---|---|---|---|
| 1 | 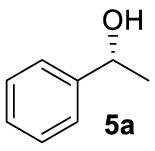 | A | >99 (86) | 6 (R) |
| 2 | 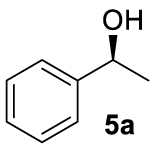 | B | 70 | 6 (S) |
| 3 | 5a | G | >99 (79) | 18 (S) |
| 4 | 5a | H | >99 (84) | 0 |
| 5 | 5a | C | >99 (92) | 34 (S) |
| 6d | 5a | C | 99 (86) | 27 (S) |
| 7 | 5a | D | 0 | |
| 8 | 5a | E | >99 (88) | 18 (S) |
| 9 | 5a | F | 91 (80) | 16 (S) |
| 10 | 5a | I | 88 (69) | 0 |
| 11 | 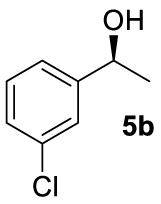 | C | >99 (95) | 16 (S) |
| 12 | 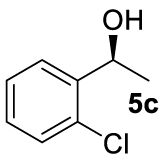 | C | 94 (75) | 10 (S) |
| 13 | 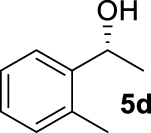 | C | 98 (89) | 8 (R) |
| 14 | 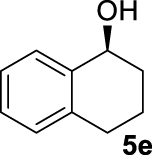 | C | >90 (88) | 20 (S) |
| 15 | 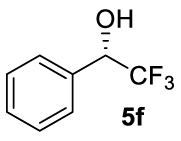 | C | >99 (90) | 4 (S) |
a Reaction conditions: ketone (1 mmol), DEMS (1.5 mmol), THF (5 mL), ZnEt2 (5 mol%), ligand (5 mol%).
b Conversion determined by 1H NMR spectroscopy after basic hydrolysis, in parenthesis, isolated yield after purification by column chromatography.
c Enantiomeric excess determined by chiral HPLC analysis.
d Reaction performed with 2.5 mol% of C in otherwise identical conditions.
At this point it seemed that a rigid bisphosphate derived from BINOL was preferable to observe the formation of enantioenriched (S)-1-phenylethanol with modest enantiomeric excesses, up to 34% ee for BINOPHAT C. The reduction of 4a in the presence of this ligand was thus further investigated by changing the solvent or the temperature: unfortunately, performing the reaction in toluene, CH2Cl2, or CH3CN, as well as decreasing the temperature in THF to 0 °C, turned out to be detrimental for the reactivity. Therefore, a series of representative acetophenone derivatives were next hydrosilylated with the combination of ZnEt2 and C in THF at room temperature. The substitution in the meta position of the aromatic ring did not affect the reactivity; indeed m-chloroacetophenone (4b) was completely converted into alcohol 5b, which was isolated in 95% yield with an ee of 16% (Table 1, entry 11). The more encumbered ortho-substituted substrates 4c and 4d also participated to the reaction leading to 5c and 5d in good yields, and in 10% and 8% ee, respectively (Table 1, entries 12 and 13). Alpha-tetralone (4e) as well was reduced to the corresponding alcohol 5e in 88% yield and 20% ee (Table 1, entry 14). Finally, the activated 2,2,2-trifluoroacetophenone (4f), which is reported to give very low enantiomeric excesses in this transformation [12, 13, 17, 20, 29], was completely converted into 5f which was recovered in 90% yield and in only 4% ee (Table 1, entry 15).
In summary, we have reported the synthesis of three new chiral BINOL-derived mono- and bisphosphotriester architectures and we have provided the proof of concept that phosphates, which are an underexplored class of ligands, can be useful in asymmetric metal-catalyzed transformations, as exemplified herein by the Zn-catalyzed hydrosilylation of prochiral ketones. It is certain that, since the application of such readily available compounds remains underexamined in the field of catalysis, our report opens up an avenue for further achievements. Indeed, the synthesis of a larger panel of ligands as well as the exploration of different reaction conditions and new catalytic transformations are underway in our laboratory and will be reported in due course.
1CCDC deposition numbers are as follow: 2012272 for G and 2012273 for H.