

1 Introduction
Cyanide-bridged bimetallic assemblies based on cyanometalate building blocks, [M(CN)m]q–, have attracted considerable recent interest in the area of molecule-based magnets [1, 2]. One of the advantages of using cyanide ligands when building magnetic materials is that they can mediate strong exchange interactions between the interconnected paramagnetic metal centres. A prominent example is the Prussian Blue family, based on the hexacyanometallates [3–9]. This family of 3d ion compounds has provided examples of magnets with critical temperatures as high as 376 K, and materials with interesting electrochemical, optoelectronic and magneto-optical properties.
Paramagnetic 4d and 5d metal ions have been envisaged only recently for the construction of molecule-based magnets [10]. Owing to the more spatially extended d orbitals for these elements, stronger exchange interactions are anticipated between magnetic centres and, consequently, magnetic ordering at higher temperatures. A synthetic approach that we currently investigate in our laboratories is based on the use of the low-symmetrical [MoIII(CN)7]4– as a building block; several two- and three-dimensional 3d–4d bimetallic compounds have been obtained [11, 12]. For instance, reaction of [Mo(CN)7]4– with Mn2+ ions permitted to obtain three cyanide-bridged bimetallic compounds exhibiting long-range magnetic ordering at Tc as high as 51 K, along with a pronounced magnetic anisotropy [13]. A higher ordering temperature is obtained when asymmetry is imposed not only to the MoIII sites but also to the MnII centre. Using triethanolamine (tea) as an additional ligand, we recently prepared the 3D bimetallic assembly [Mn2(tea)Mo(CN)7], which exhibits long-range magnetic ordering bellow Tc = 106 K [14].
We further reasoned that, using Co2+ instead of Mn2+, it should be possible to enforce a higher magnetic coercitivity for the material. Surprisingly, the reaction of [Mo(CN)7]4– with CoX2 (X = nitrate or perchlorate), under strictly anaerobic conditions, did not afford the expected compound, but crystals of the 3D bimetallic assembly [{Co(H2O)2}2Mo(CN)8]·4 H2O, whose structure and magnetic properties are reported here.
2 Results and discussion
2.1 Preparation and structural features
The reaction of K4[Mo(CN)7]·2 H2O with [Co(H2O)6]X2 (X– = NO3– or ClO4–) in a slow diffusion process of aqueous solutions affords the 3–D cyanide-bridged bimetallic compound of formula [Co2(H2O)4Mo(CN)8]·4 H2O, 1. Orange octahedral crystals of 1 are formed reproducibly in an H-shaped tube from deoxygenated solutions of reagents. A rational preparation for compound 1 starting from [MoIV(CN)8]4– and [Co(H2O)6]2+ was reported by McKnight et al. [15]. However, following the described synthetic procedure, a powdered sample is formed and the crystal structure of the compound remained unknown.
The single-crystal X-ray structural determination revealed that 1 has a polymeric structure based on a network of [Mo(CN)8] units and [CoII(H2O)2] ions. The axial-coordinated water molecules point directly into the channels of the network and are involved in H–bonding interactions with H2O solvated molecules located in the void of the framework (Fig. 1). The geometry of the Mo(CN)8 unit is close to a square antiprism. The CoII ion is six-coordinated with four cyanide-nitrogen and two water-oxygen atoms forming a distorted octahedral environment. The coordination sphere for each metal centre and their interconnections are depicted in Fig. 2 with selected bond lengths and distances. Each Mo(CN)8 building block is linked to CoII ions through its eight CN ligands and each CoII centre is connected to four Mo units forming a three-dimensional framework. The organization is highly symmetrical and may be described as a grid-like 3D arrangement of Co←NC–Mo–CN→Co linkages, an arrangement reminiscent to that found in related [M(CN)8]4–/M2+ 3D–coordination polymers [16–19].
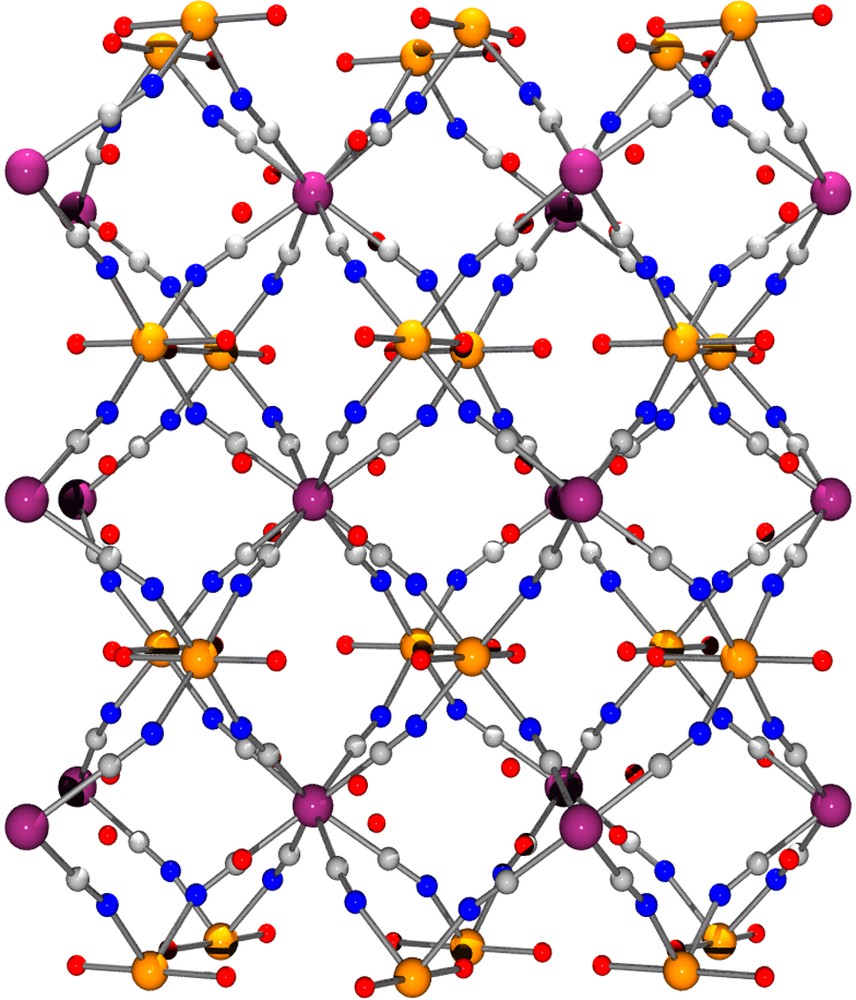
View of the 3D arrangement of [{Co(H2O)2}2Mo(CN)8]·4 H2O down the b direction.
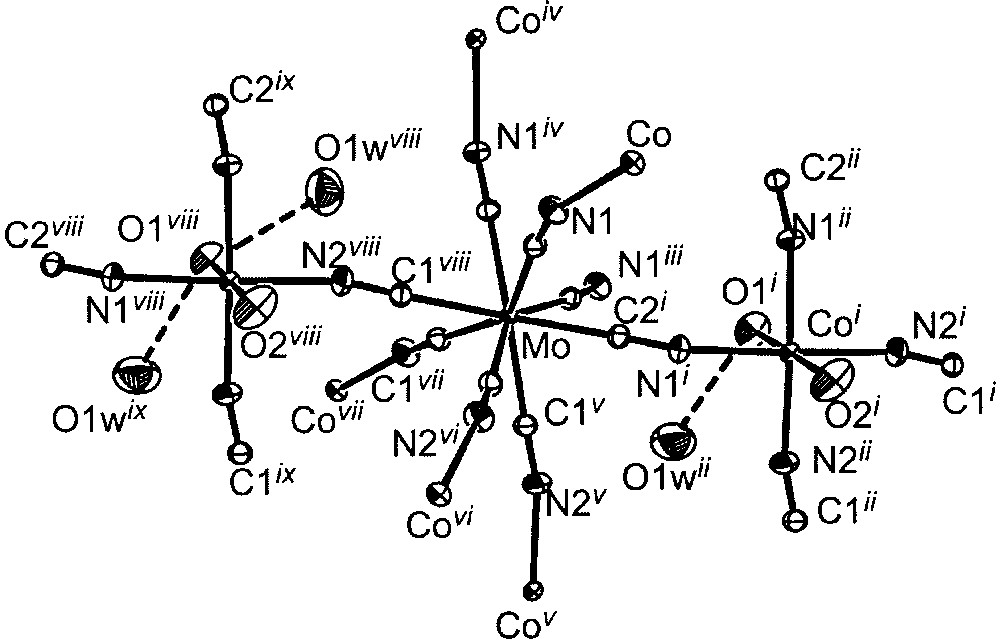
ORTEP view of the elementary fragment with labelling scheme for [{Co(H2O)2}2Mo(CN)8]·4 H2O showing the coordination spheres of the metal ions (ellipsoids at 30% probability level). Selected bond lengths (Å) and angles (°): Mo–C1, 2.162(2); Mo–C2, 2.158(2); Co–N, 2.105(2) to 2.140(2); Co–O, 2.083(3) to 2.117(3); O1w···O1, 2.875(4); O1w···O2, 3.016(4); Mo–C–N, 174.6(2) and 175.8(2); Co–N–C, 156.0(2) and 166.7(2). i: –y, x, –z; ii: –y, x, –z; iii: –x, –y, z; iv: y, –x, z; v: y + 0.5, –x + 0.5, z + 0.5; vi: –x + 0.5, –y + 0.5, z + 0.5; vii: x + 0.5, y + 0.5, z + 0.5; viii: –y + 0.5, x + 0.5, z + 0.5 ix: –y + 0.5, x + 0.5, – z + 0.5.
This coordination scaffold is framing square-shaped apertures forming channels where both the coordinated H2O and no-coordinated molecules are located. A view of the framework along the c direction depicted in Fig. 3 illustrates this feature. The aperture of a channel is delimited by four Co atoms defining a 5.93 (1.87) × 6.06 (2.0) Å parallelogram (the value between brackets takes into account the van der Waals radii of 2.03 Å for Co), the diagonal separations being 6.54 (2.48) Å and 10.05 (5.99) Å. This opens a cavity whose section is defined by the Mo ions arranged in a square with sides of 8.295 (3.755) Å (van der Waals radii for Mo = 2.27 Å), the diagonal distance being 11.731 (7.19) Å; its length, i.e. the Co to Co distance, is 6.75 (2.69) Å. The succession of cavities constitutes the channels.
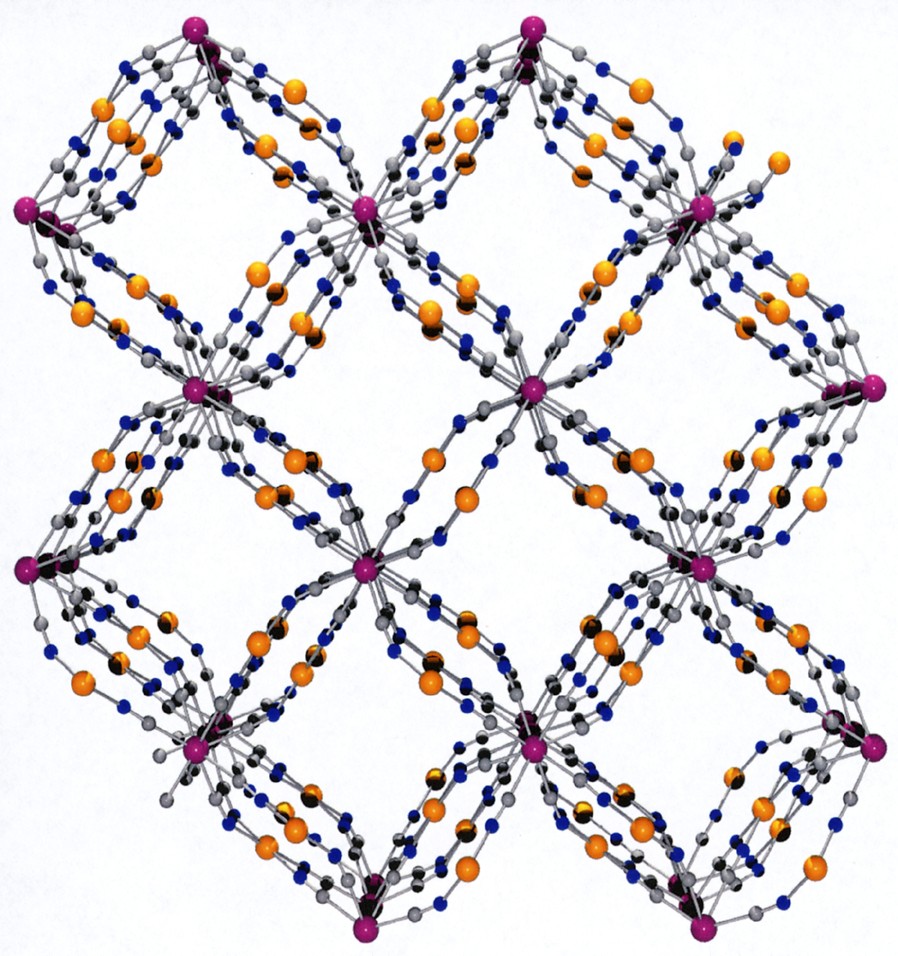
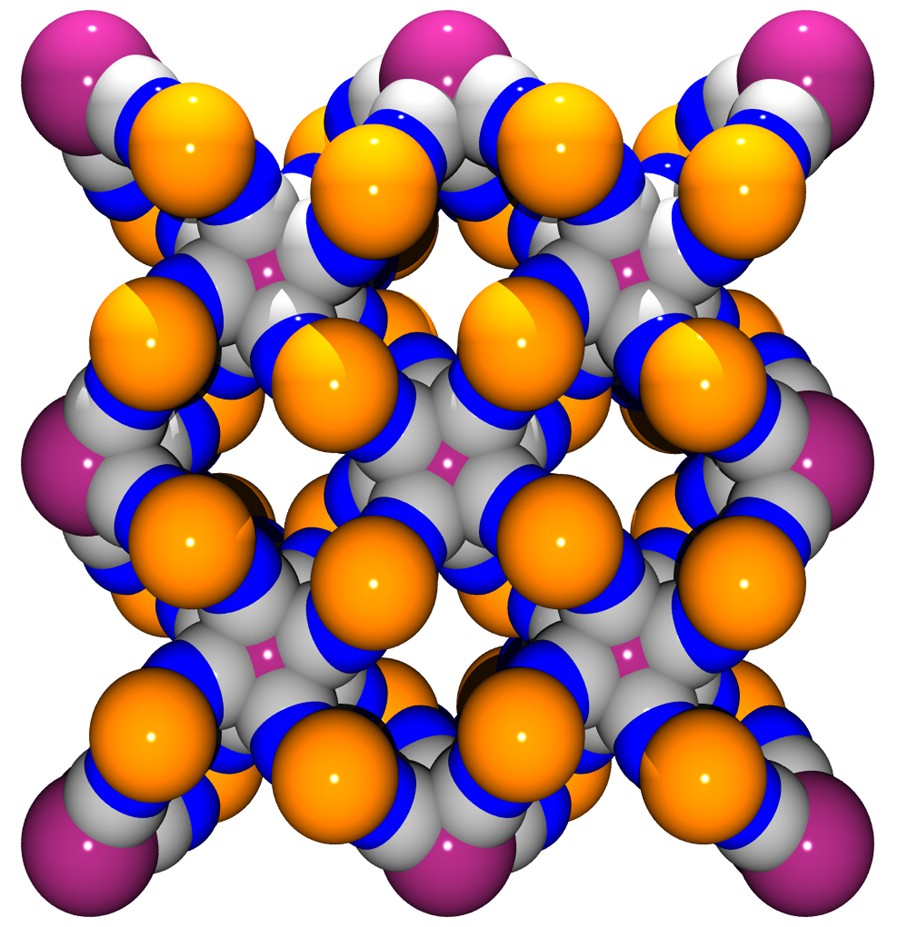
(a) View along c of the framework without the H2O molecules highlighting the channels running through the structure. (b) Space-filling model view.
2.2 Spectroscopic characteristics
The IR spectrum of the crystalline compound shows only one νCN band centred at 2138 cm–1, supporting the formation of MoIV–CN–CoII linkages [15, 17]. Two broad bands centred at 3554 and 3406 cm–1 are due to O–H stretching modes and support the presence of both coordinated and non-coordinated water molecules in 1. The diffuse reflectance electronic spectrum shows the superposition of characteristic bands of the [MoIV(CN)8]4– (380 and 430 nm) and CoII (490 nm) fragments [20]. No additional absorption that could be attributed to metal-to-metal charge-transfer bands was observed.
2.3 Magnetic properties
The temperature dependence of the molar magnetic susceptibility, χM, of compound 1 was measured on a polycrystalline sample in the temperature range 5–300 K using a SQUID magnetometer. The result, plotted as χM T versus T in Fig. 4, shows that the value for χM T decreases from 5.9 cm3 K mol–1 at 300 K to 3.65 cm3 K mol–1 at 5 K. The decrease of χM T with T can be attributed to the intrinsic behaviour of CoII rather than to antiferromagnetic exchange interactions among these ions in the 3D network. This is confirmed by the field dependence of the magnetization recorded at 2 K, which reaches a saturation value of 4.25 μB for 5 T (Fig. 4, insert).
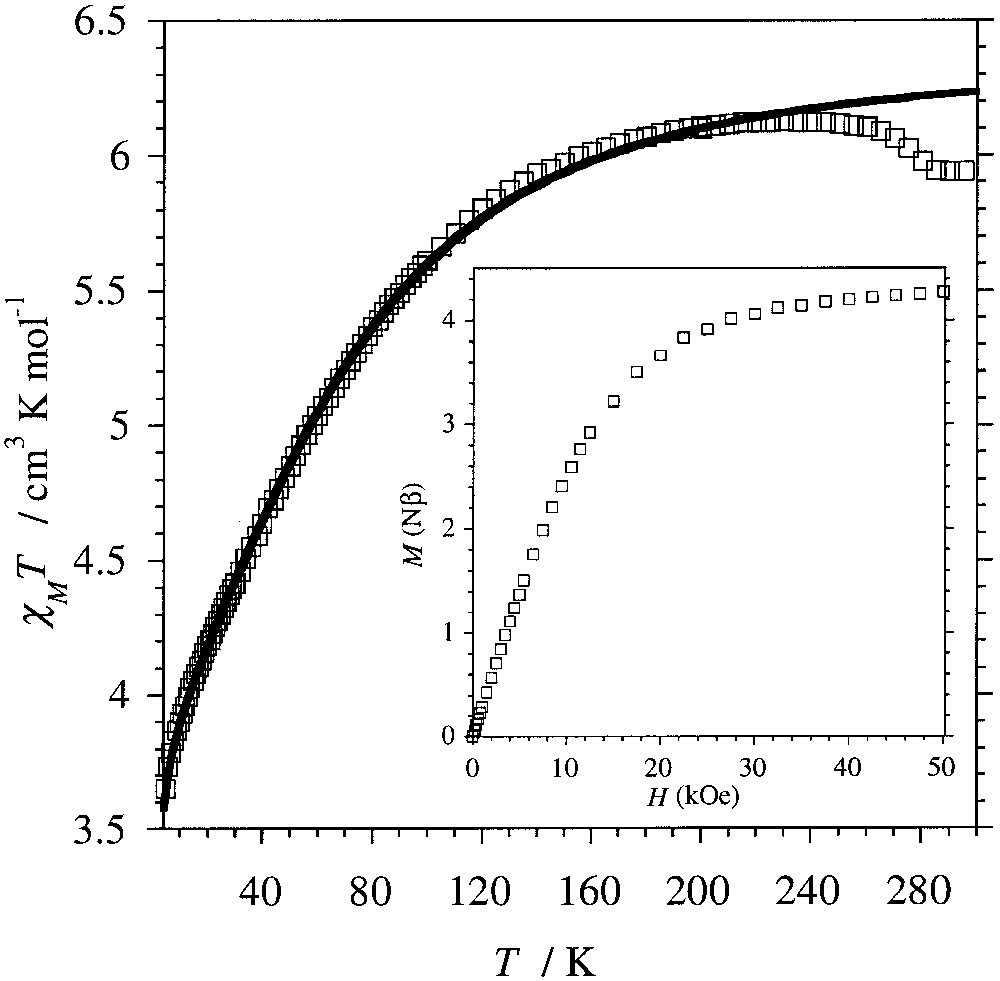
Temperature dependence for the experimental (□) and calculated (–) χM T for [{Co(H2O)2}2Mo(CN)8]·4 H2O yielding g = 2.604 ± 0.001, D = –80.3 ± 0.6 cm–1 and θ = –0.36 ± 0.01 K. Insert: Field dependence of the magnetization recorded at 2 K.
The high-temperature χM T value obtained for compound 1 is much larger than the spin-only value expected for two S = 3/2 spins (3.75 cm3 K mol–1). This deviation is the result of the first-order orbital momentum displayed by high-spin CoII ions in octahedral surrounding. Moreover, in addition to the symmetry lowering due to the deformation of the octahedral coordination sphere of the Co ions, the coupling of the first-order orbital momentum with the spin momentum partially removes the degeneracy of the ground state and the excited states. This results in an energy spectrum for a high-spin CoII ion where the two low-lying levels correspond to the MS = ±1/2 and ±3/2 states. Taking into account the Zeeman perturbation resulting from the applied external field, the Hamiltonian describing this situation is [21]:
This expression includes the energy gap between the states split by the deformation of the coordination sphere, Δ, a spin-orbit coupling parameter, λ, and a parameter κ, called the orbital reduction factor accounting for the effect of the M–ligand bond on the orbital momentum. A model taking into account all these parameters to fit the experimental magnetic data would be over-parameterised. Therefore, it is usual to consider that Δ is large enough to have only the two lowest Kramer doublets thermally populated in the investigated temperature range. The energy separation D between these levels is then considered as a zero-field splitting within the quartet state. The Hamiltonian considered in that case becomes H = D [Sz2 – S (S + 1)/3] + β g·S·H. The orbital momentum does not longer appear but its influence is incorporated into both D and g parameters.
The theoretical expression for the molar susceptibility deduced for this spin Hamiltonian is given in eq. (1) [22]. It considers the paramagnetic behaviour for the two independent CoII ions found in the molecular formula. Possible weak interactions between the Co(II) sites within the network are considered through a Curie–Weiss parameter θ.
| 1 |
Best fit to the experimental data in the temperature domain 5–250 K leads to gCo = 2.604 ± 0.001, D = –80.3 ± 0.6 cm–1 and θ = –0.36 ± 0.01 K (Fig. 4). The small θ value is consistent with weak antiferromagnetic interactions between the Co(II) ions through the diamagnetic NC–MoIV–CN bridges. Such a weak exchange through this spacer has been observed previously in related compounds with MnII and CuII instead of CoII ions [18]. The data above 250 K have not been considered for the parameterisation, because of the small step visible close to 270 K. This discontinuity in the magnetic behaviour is attributed to a slight distortion of framework occurring for these temperatures.
An analysis of the field dependence of the magnetization with the Brillouin function by considering gCo = 2.60 indicates that at 2 K the effective spin for the CoII ions in compound 1 is still above S = .
Both the spectroscopic data and the magnetic study are consistent with a Mo ion in oxidation state IV. Thus, in the course of the crystallization process, the 4d metal ion is oxidized from MoIII in [Mo(CN)7]4– to MoIV in the diamagnetic [Mo(CN)8] building block of the framework. The mechanism for this oxidation process remains unclear, but the propensity for [Mo(CN)7]4– to easily be oxidized to [Mo(CN)8]4– has been reported before [23]. An analogous oxidation process was observed in a related self-assembling procedure involving [Mo(CN)7] and an MnII compound [24]. In both cases, the reactions were performed under strictly anaerobic conditions and no other compound could be detected in the solid state.
3 Concluding remark
The evolution of [Mo(CN)7]4– during the slow crystallization process in the presence of CoII ions leads to the formation of single crystal of [{Co(H2O)2}2Mo(CN)8]·4 H2O. This provided the opportunity to investigate the crystal structure for this compound, revealing its 3D nature. Interestingly, this compound exhibits a porous framework with square-shaped channel running through the structure. Its magnetic properties are mainly governed by the zero-field splitting due to the Co ions.
4 Experimental
The commercial chemicals were used as received without further purification. K4[Mo(CN)7]·2 H2O was synthesized as described in the literature [18]. All physical measurements were performed on macroscopic crystals ground to a powder. Infrared spectra (400–4000 cm–1) were recorded from KBr pellets on a Perkin-Elmer Paragon 1000 FTIR spectrometer. UV–Vis spectra (diffuse reflectance technique) were recorded with a UV–4 (Unicam) spectrophotometer. Variable-temperature (2–300 K) magnetic susceptibility measurements were carried out with a Quantum Design MPMS–5S SQUID magnetometer under an applied magnetic field of 5000 G. Diamagnetic corrections were estimated from Pascal tables and magnetic data were corrected for diamagnetic contributions of the sample holder.
4.1 Synthesis
Orange octahedral crystals of [{Co(H2O)2}2Mo(CN)8]·4 H2O were obtained by slow diffusion, in an H-shaped tube (V = 60 ml) under nitrogen, of two deoxygenated 5-ml aqueous solutions containing 35 mg (0.075 mmol) of K4[Mo(CN)7]·2 H2O and 43 mg (1.5 mmol) of Co(NO3)2·6 H2O, respectively. The crystals were carefully collected, washed with water and ethanol, and finally dried under a flow of N2. Yield: 30–35%.
A second sample generated by slow interdiffusion of two aqueous solutions of K4[Mo(CN)7]·4 H2O (55 mg) and Co(ClO4)2.6H2O (60 mg) in an H-tube, proved to correspond to the same compound.
Elem. anal.: calcd for C8H16N8O8Co2Mo: C, 16.97, H, 2.85; Co, 20.82; Mo, 16.95; N, 19.79%; found: C, 17.00; H, 2.81; Co, 20.83; Mo, 16.88; N, 19.68%. Selected IR data (KBr, cm–1): 3554, 3406, 2138, 451, 420.
4.2 Solution and refinement of the X–ray structure
Crystal data and structure refinement of [{Co(H2O)2}2Mo(CN)8]·4 H2O, 1 are given in Table 1.
Crystal data and structure refinement of [{Co(H2O)2}2Mo(CN)8]·4 H2O, 1.
| Empirical formula | C8H16Co2MoN8O8 |
| Formula weight | 566.09 |
| Temperature (K) | 293(2) |
| Crystal system | Tetragonal |
| Space group | I4/m |
| a (Å) | 11.7306(4) |
| c (Å) | 13.0372(5) |
| U (Å3) | 1794.02(11) |
| Z | 4 |
| Dc (g cm–3) | 2.096 |
| m (mm–1) | 2.573 |
| Reflections collected | 4034 |
| Independent reflections | 2479 |
| Parameters | 67 |
| Goodness-of-fit on F2 | 1.039 |
| Final R indices | R1 = 0.0434 |
| [I > 2 s(I)] | wR2 = 0.0860 |
| R indices (all data) | R1 = 0.0699 |
| wR2 = 0.0964 |
A single crystal of the title compound was mounted on glass fibre and transferred to a Kappa Nonius four circle diffractometer equipped with a CCD camera and a graphite monochromated Mo Kα radiation source (λ = 0.71073 Å). Data were collected at room temperature, then, treated using DENZO and SCALEPACK [25], no absorption correction were performed. Crystal of [Co(H2O)2]2[Mo(CN)8]·4 H2O crystallizes in the tetragonal space group I4/m, a = b = 11.730 6(4) Å, c = 13.0372(5) Å, V = 1794.0(1) Å3, Z = 4. Data were refined with SHELXL–97 [26] program by full matrix least squares method, on F2; refinement of variables were performed with anisotropic thermal parameters for all non-hydrogen atoms. 4034 reflections (completeness to 37.74° = 99.6%) were collected giving rise to Rint = 0.0411. Refinements of 67 parameters gave R1 = 0.0434 and wR2 = 0.860 for 1847 data ≥ 2 σ(I) and R1 = 0.0699, wR2 = 0.0964 for 2479 unique data. No H atoms from water molecules were included in this model, the residual electron-density being in the range –1.316 to +1.580 e Å–3 with the largest peak trough close to the oxygen atoms from iron-coordinated water molecules. Despite to these peaks, and contrary to a related structure [19], no disorder between coordinated water molecules is observed.
5 Supplementary material
The supplementary material has been sent to the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge, CB2 1EZ, UK as supplementary material No. 195769 (CIF) and can be obtained by contacting the CCDC (fax: +44–1223–336033; e-mail: deposit@ccdc.cam.ac.uk or www: http://www.ccdc.cam.ac.uk).
Acknowledgements
This work was supported by the French Ministry of Research (fellowship to FT) and the TMR Research Network of the European Union entitled ‘Molecular Magnetism: from Materials toward Devices’.