

1 Introduction
Chirality plays an essential role in all the fields of chemistry. Many significant compounds (amino acids, peptides, carbohydrates, terpenes, alkaloids, pharmaceutical compounds...) are optically active and the respective R and S enantiomers may have a very different biological activity. Consequently, in order to perform the characterization of natural or synthetic products, the analysis of chiral compounds took a great importance and represents a challenge for the chemists. Among the methods allowing the chiral distinction, mass spectrometry experienced these last years a significant development with chemical ionisation [1] (CI), fast atom bombardment [2] (FAB), electrospray [3] (ESI) and also recently with matrix assisted laser desorption/ionisation (MALDI) [4].
Carbohydrates are involved in a host of biological functions, including cell–cell recognition, cell–cell interaction and molecular targeting. The structural elucidation of the carbohydrates is the first step to understand how and why they assist these biological functions. This purpose requires knowledge of the monosaccharide composition and sequence, linkage position, and branch points, as well as anomeric configuration of each glycosidic bond. For carbohydrates, several approaches may be used to contribute to their structural elucidation [5]. Distinction of diastereomeric monosaccharides is often a tedious task by mass spectrometry, and ultimately the monosaccharide stereochemistry must be determined. A rapid and efficient method to differentiate four diastereoisomeric saccharides (M = glucose, mannose, galactose and talose) (Fig. 1) in the presence of iron (II) metal ion under electrospray (ESI) conditions has been described in a previous work [6].

Structure of the various studied hexoses.
The approach was based on ion neutral interaction between the sugar analyte and the iron (II) ion. Differentiation was made from the dissociation of ternary [MFeIICl]+ species. The daughter ions of these clusters were produced using the resonant excitation mode and their respective formations are characterized by different rate constants related to the hydroxyl stereochemistry. It is possible to distinguish stereoisomeric sugars by examining the change of the relative abundance of the diagnostic fragment ions. When the sugar molecule (M) is introduced in the ESI source, in excess with the iron (II) chloride, the formation of doubly charged multimeric M(n)Fe2+ ions is observed. The behaviour of these cluster ions under resonant excitation conditions reveals an increase of the stereochemical effects compared with that observed in the case of singly-charged monomeric [MFeIICl]+ ions. The orientation of the dissociation pathways of doubly charged multimeric M(n)Fe2+ complexes appeared to depend on the monosaccharide n number. This represents an advantageous alternative for the structural elucidation of their respective stereochemistry.
It has been shown [7] that the halogen (X) has a significant effect on the extent of diastereomeric differentiation observed after the collision-induced dissociation (CID) of cationized monomeric [MFeIIX]+ and dimeric [M2FeIIX]+ ions. By scrutinizing the relative abundances variation of the diagnostic fragment ions, it is possible to distinguish these stereomers. An enhancement of the stereochemical effects was observed from the CID of cationized dimeric [M2FeIICl]+ complexes [8]. Nonetheless, in all cases, it has been possible to distinguish the diastereomers. The differentiation was also obtained from [MM′FeIICl]+ cationized heterodimers with M′ corresponding to a diastereomeric diol. The production of the latter heterodimers is very useful for the distinction of position isomer diols like associations of monosaccharide isomers, α/β anomers, in the presence of propanediol, butanediol or pentanediol isomers.
The study of these cationized heterodimers under collision dissociation conditions allowed us to go further and use natural sugars (optically pure D series) to distinguish chiral molecules [9]. In chemical ionisation (positive mode), chiral reagents allowed a long time ago the distinction of the enantiomers of tartric acid [10], borneols [11], cyclic aminoacids and esters [12]. More recently, Cooks [13] has used the negative ion mode to differentiate butanediol enantiomers, from the dissociation of non-covalent and covalent heterodimers. Alternatively, using aminoacids our group used heterochiral bi- and ternary-coppered complexes to distinguish amino acids [14]. In this present study, we explore and demonstrate the capacity and the efficiency of natural sugars (optically pure D series) to distinguish diol diastereomers and position isomers and extend the method to chiral molecules using Fe(II) transition metal.
2 Results and discussion
2.1 Distinction of diastereoisomers from cationized dimers
2.1.1 Distinction of cis- and trans-1,2-indanediol by their respective reactivity with monocarbohydrates
The differentiation of the stereochemistry between cis- and trans-1,2-indanediol (Fig. 2) is particularly difficult with a conventional ion source. Few successful results were indeed obtained in positive chemical ionisation with ammonia via a SN2 or SNi nucleophilic substitution process [15] or with dimethylether [16]. With ammonia, substituted [M + NH4–H2O]+ ions are formed [17], which are studied by reactive collisions with methylamine [18] in the collision cell of a triple quadrupole instrument. Collision-activated-reaction (CAR) spectra provided additional information concerning the structure of the product species using very low collision energy. In practice, it is particularly difficult to obtain reproducible results.
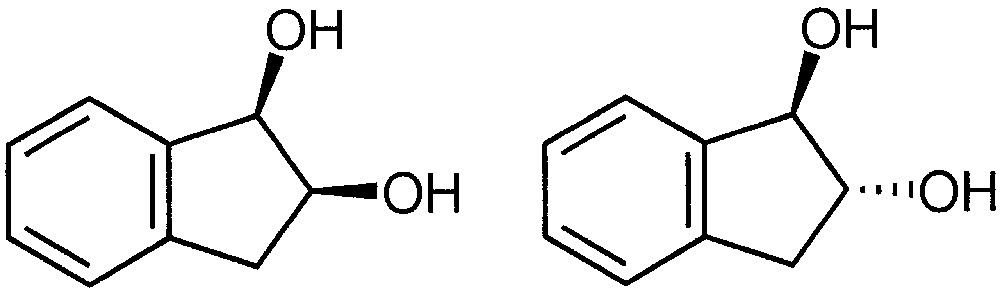
Structure of cis- and trans-1,2 indanediol.
Under ESI conditions, it is very simple and efficient to prepare cationized heterodimers in the gas phase from solutions containing cis- or trans-indanediol (Indc/Indt) and a monosaccharide (M) in equimolar mixture together with an excess of FeCl2. The ions observed in the ESI mass spectra correspond to (i) cationized [Glu,FeIICl]+ monomers (m/z 271) and cationized [Glu2FeIICl]+ dimers (m/z 451) for glucose (noted Glu), (ii) cationized monomers [Indc/Indt, FeIICl]+ (m/z 241) as well as cationized [Indc/Indt,FeIICl]+ dimers (m/z 391), and (iii) cationized heterodimers [Indc/Indt,GluFeIICl]+ (m/z 421). When this experiment was repeated with the others stereoisomeric sugars, similar multimeric species were detected (non-reported results).
The cis or trans stereochemistry of 1,2-indanediols is efficiently characterized by the CID spectra of the hetero multimeric [Indc/Indt,M,FeIICl]+ (m/z 421) complex ions as shown in Fig. 3. Strong variations on the ions relative abundance are displayed on the CID spectra using mannose (Fig. 3a and b) and galactose (Fig. 3c and d). The same behaviour was observed using glucose and talose (data not shown). The collision spectra of the cationized monomers [IndcFeIICl]+ and [IndtFeIICl]+ were identical due to the prompt elimination of H2O from the leaving hydroxyl group resulting into the loss of the cis/trans stereochemistry.

Product ion scan of the cationized heterodimers [MM′ + FeII + Cl]+ (m/z 421) (a) cis-indane + mannose, (b) trans-indane + mannose, (c) cis-indane + galactose, and (d) trans-indane + galactose recorded using an excitation amplitude of 0.8 Vp–p.
From these CID spectra, it appears that the competitive losses of HCl (m/z 385) and Indt/Indc partners are influenced by the cis/trans diastereochemistry of indane diols when galactose is used compared to that observed with mannose. Interestingly, the cis/trans differentiation is reinforced when an axial group is at C(4) position for galactose rather than at C(2) for mannose. Alternatively from mannose, the loss of water as well as those of 60 u (i.e., C2H4O2) and 96 u (i.e., C2H4O2 + HCl) is reinforced from trans isomer, although stability of precursor m/z 421 is similar (i.e., close relative abundance from cis/trans isomers).
2.1.2 Distinction of α- and β-methyl-glucose by their respective reactivity with glycerol
From a mixture of α/β-methyl-glucose (α/βGlu), glycerol (Gly) and FeCl2 various cationized species are observed in the mass spectra. Among these species, [(Gly,α/βGlu)FeIICl]+ m/z 377 (100%) is displayed. On a lower extent, another deprotonated/cationized species [(Gly,α/βGlu)–H + FeII]+ m/z 341 (20%) is observed. These ions were studied under CID conditions. Fig. 4a and b show MS/MS spectra of [(Gly,αGlu)FeIICl]+ and [(Gly,βGlu)FeIICl]+ (m/z 377), respectively, which display some differentiation of these anomeric species from the extent of the relative abundance of the ions produced from direct competitive decomposition. The loss of HCl leads to m/z 341 competitively with the loss of glycerol leading to the cationized monomer [α/βGluFeIICl]+ m/z 285. Nevertheless, few ions contribute to differentiation in this case. On the other hand, the MS/MS spectra of the deprotonated/cationized species m/z 341 (Fig. 4c and d) display important stereochemistry effect through numerous ions. Indeed, the precursor m/z 341 ions lead to m/z 309, corresponding to the loss of MeOH. All the other ions correspond to consecutive and competitive decomposition of m/z 309, as confirmed by MS3 experiments (data not shown). Noteworthy, the loss of H2O (m/z 291) followed by the elimination of 60u (C2H4O2) leading to m/z 231 is observed. In the same way, the elimination of 60 u (C2H4O2), 90 u (C3H6O3) and 102 u (C4H6O3) lead to m/z 249, m/z 219 and m/z 207, respectively. Similar results were obtained using α/β-methyl-galactose. Noteworthy, the absence of Cl– in the complex leads to strong modifications in the CID spectra compared to that of [(α/β-Glu, Gly)-H + FeII]+. Likely, the loss of glycerol from [(α/β-Glu, Gly)-H + FeII]+ should be favoured because of its lower acidity (compared to glucose). Nevertheless, because of the absence of Cl– counter-ion, the elimination of one ligand from FeII is more difficult. Consequently, the direct elimination of glycerol (m/z 249) is not the major process. In fact, this ternary complex prefers to dissociate by covalent C–C and C–O bond cleavage rather than release either the monosaccharide (absence of m/z 161) or glycerol (low abundance of m/z 249). In fact, the loss of CH3OH (m/z 309) and the consecutive (CH3OH + H2O) losses (m/z 291) are significantly favoured from the α-methyl glucose as well as the formation of m/z 207 (loss of C4H6O3). In all cases, presence of iron is preserved in the ions and then, neither eliminate during precursor ion dissociation. Interestingly, both the H2O and C4O3H6 losses are significantly influenced by the initial stereochemistry of the anomeric site. Although its elimination was required for consecutive dissociations, the mechanism of this process is in progress.

Product ion scan of the cationized heterodimers [glucose + glycerol + FeII + Cl]+ (m/z 377) (a) α-glucose, (b) β-glucose β-glucose recorded using an excitation amplitude of 0.7 Vp–p and of deprotonated/cationized heterodimer [glucose + glycerol–H + FeII]+ (m/z 341), (c) α-glucose and (d) β-glucose recorded using an excitation amplitude of 1.2 Vp–p.
2.2 Distinction of enantiomers from cationized dimers using (R,R)- and (S,S)-2,3-butanediol
These results on diastereomeric diols and triols was applied for distinguishing enantiomeric diols such as (R,R)-2,3-butanediol and (S,S)-2,3-butanediol (Fig. 5).
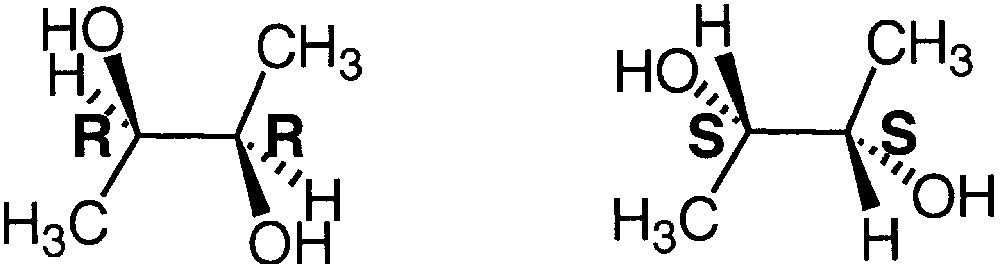
Structure of (R,R)-2,3-butanediol and (S,S)-2,3-butanediol.
For that purpose, cationized heterodimers [MM′FeIICl]+, with M′ constituted by (R,R)- or (S,S)-2,3-butanediol (respectively noted But(R,R) and But(S,S)) are formed with optically pure underivatized monosaccharides M (α-β-methyl-glucose, α-β-methyl-galactose).
The ESI mass spectra, obtained from a stoichiometric mixture of β-methyl-galactose and (R,R)-2,3-butanediol (with FeCl2 in excess) is presented in Fig. 6. Many cationized monomeric or dimeric ions issued from (R,R)-2,3-butanediol and β-methyl-galactose are displayed. Among these species, several cationized heterodimers such as [MM′FeIICl]+ ([(But(R,R),βGal)FeIICl]+, m/z 375) and [(MM′)–H + FeII]+ ([But(R,R),βGal–H + FeII]+, m/z 339) are observed for instance, but in a relatively weak abundance.
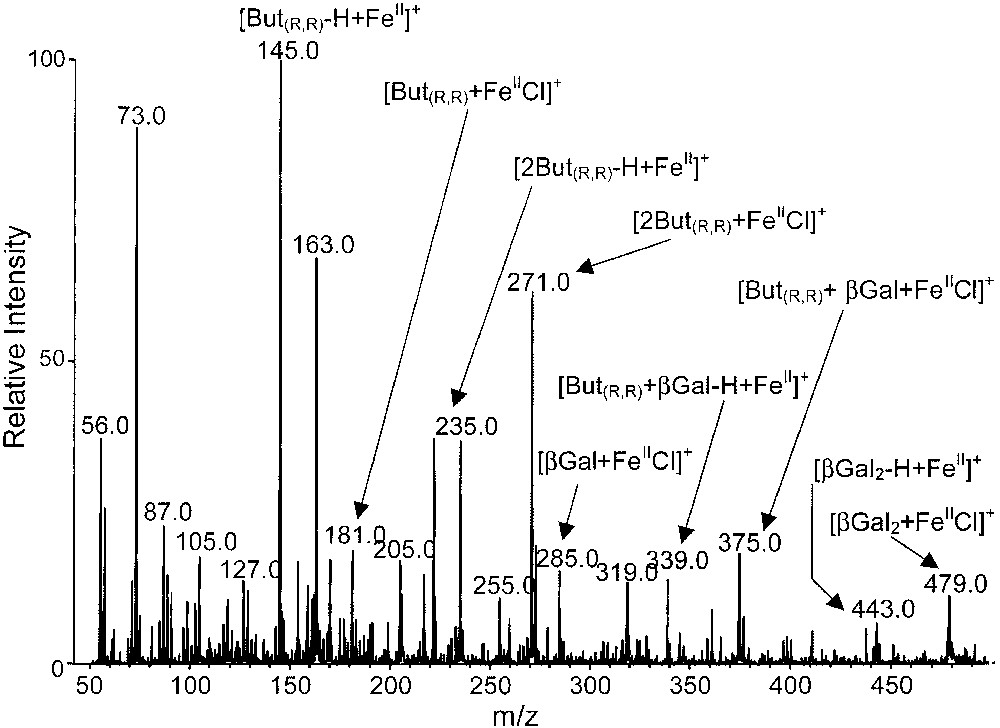
ESI mass spectrum of a stoichiometric mixture of β-methyl-galactose and (R,R)-2,3-butanediol with FeCl2 (in excess).
The CID spectra of the cationized [MM′FeIICl]+ heterodimers did not allow any significant chiral differentiation of the 2,3-butanediol enantiomers, unlike the case of diastereoisomers (data not shown). However, a spectacular enantiomeric differentiation was observed from the CID spectra of the cationized deprotonated [(MM′–H) + FeII]+ dimeric ions. The best results were provided from the cationized heterodimers constituted by α or β methyl-glucose (Fig. 7) and by α and β methyl-galactose (Fig. 8).

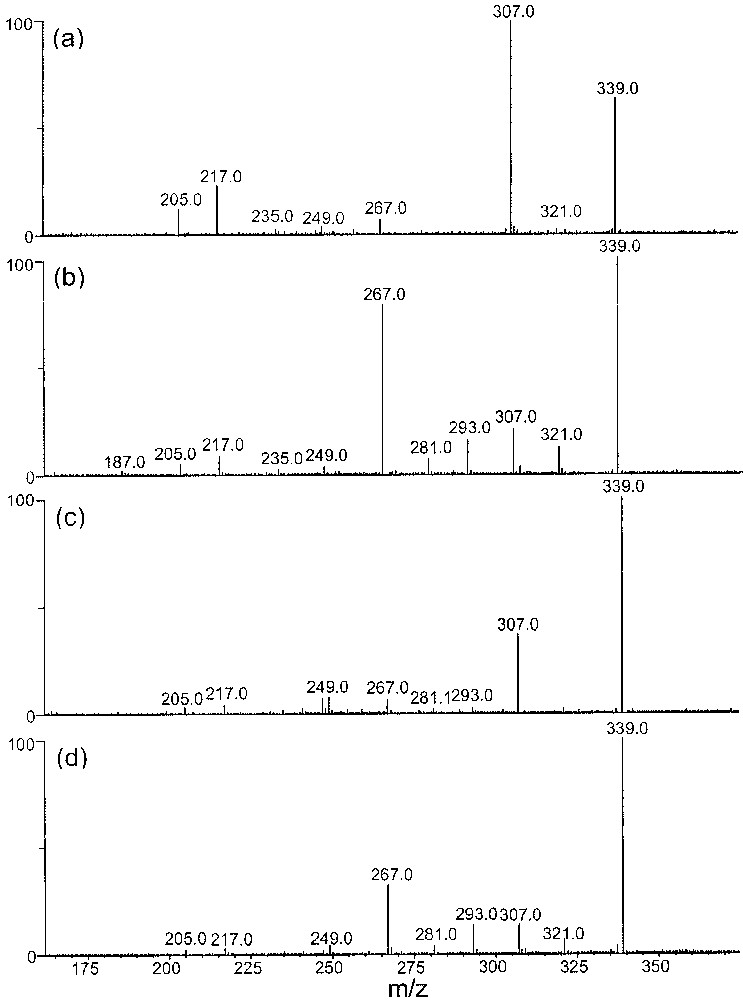
CID spectra of [(MM′–H) + FeII]+ (m/z 339) heterodimers recorded using a resonant excitation of 1.2 Vp–p, with (a) M′ = (R,R)-2,3-butanediol, M = α-methyl-glucose, (b) M′ = (S,S)-2,3-butanediol, M = α-methyl-glucose, (c) M′ = (R,R)-2,3-butanediol, M = β-methyl-glucose, and (d) M′=(S,S)-2,3-butanediol, M = β-methyl-glucose.
CID spectra of [(MM′–H) + FeII]+ (m/z 339) heterodimers recorded using a resonant excitation of 1.2 Vp–p with (a) M′ = (R,R)-2,3-butanediol, M = α-methyl-galactose, (b) M′ = (S,S)-2,3-butanediol, M = α-methyl-galactose, (c) M′ = (R,R)-2,3-butanediol, M = β-methyl-galactose, and (d) M′ = (S,S)-2,3-butanediol, M = β-methyl-galactose.
Independently of the used anomeric methoxy-monosaccharide, the CID spectra presented in Figs. 7 and 8 allow to differentiate without any ambiguity both the (R,R)- and (S,S)-2,3-butanediols by the comparison of the relative abundance of the product ions. The main ions observed on the CID spectra correspond to the direct competitive loss of H2O (m/z 321) and MeOH (m/z 307) from the parent ion. Their origin is difficult to determine as they may come either from the sugar or from the chiral diol. The m/z 289 ions are exclusively present with α/β methyl-glucose. The loss of 72 u (m/z 267) and 46 u (m/z 293) probably comes from the chiral diols, because these two fragmentations are never observed in the CID spectra of cationized deprotonated homodimers (m/z 443) of methyl-saccharides. From sequential MSn experiments, the m/z 307 ions lead to m/z 217 ions by the loss of 90 u (C3H6O3) and m/z 205 by the loss of 102 u (C3H4O2). Such eliminations have been observed in the CID spectra and confirm their respective origin.
Among the various features useful for distinguishing the RR/SS enantiomers independently to the chiral partner (i.e., α-β-methyl-glucose, α-β-methyl-galactose), the following ratios [m/z 307]/[m/z 321], [m/z 307]/[m/z 267] and [m/z 289]/[m/z 293] are reinforced for the RR epimer. Both the former ratios compare the loss of methanol (m/z 307) to that of 72 u (m/z 267) and of 18 u (m/z 321), which are direct eliminations occurring from the selected deprotonated cationized complex. The loss of water may take place competitively from both the organic species constituting the complex while the loss of 72 u (C3H6O2) come from the butanediol moiety, as it was not observed from the complex constituted by glycerol and α-β-methyl-glucose/galactose.
3 Conclusion
In this study, a new method has been described to differentiate diastereoisomers or enantiomers using dimeric species constituted by a monosaccharide and a polyol cationized by FeII under ESI conditions in the gas phase. The formation of cationized [MM′FeIICl]+ heterodimers has revealed itself very easy and useful. The resonant excitation of cationized heterodimer [MM′FeIICl]+ appears to be an efficient approach to differentiate diol diastereomers. α/β-methyl galactose anomers were also differentiated from the same cationized heterodimer, but better results were obtained using deprotonated cationized heterodimer [(MM′–H)FeII]+. Finally, the distinction of enantiomers was possible only using deprotonated [(MM′–H)FeII]+ heterodimers. The CID spectra of these species showed spectacular enantiomeric effects when comparing the relative fragment ion abundances. Using sequential MSn experiments, the origin of these product ions from direct competitive and consecutive dissociations was determined. A model, based on steric interactions and ferrous cation coordination, is currently under work. Gas phase reactivity of other enantiomers is currently investigated, such as amino alcohols, terpenols and synthetic chiral alcohol and amine.
4 Experimental
The ESI mass spectra and sequential MSn experiments were performed in an ion-trap mass spectrometer (ESQUIRE, Bruker, Bremen, Germany) equipped with an Analytica of Brandford (Brandford, CT, USA) external electrospray source. Analysed solutions were prepared in a 1:1 mixture of acetonitrile/water containing monosaccharide, diol and FeCl2 in an 1:1:5 ratio. Dilution was done in order to achieve a final sugar concentration 2.5 × 10–5 M. These solutions were introduced into the external electrospray source by direct infusion using a syringe pump (120 μl h–1). The solution samples were flowed through a grounded stainless steel capillary at atmospheric pressure and were electrosprayed across a 4-kV potential through heated nitrogen drying gas assisting the spraying process. Ionisation/desorption conditions were optimised for providing abundant cationized molecules such as the [MM′FeIICl]+ and [(MM′-H)+FeII]+ heterodimers within a great sensitivity for performing reproducible sequential MS/MS experiments. The m/z ratio range of analytical scan was from 50 Th to 2000 Th (standard scanning mode). The resonant ion ejection occurs at βz = 2/3 (qz = 0.78) according to the m/z ratio resolution of 0.4 Th (within scan rate of 2000 Th s–1). All ESI mass spectra were recorded on positive mode with a capillary voltage, 4 kV; end plate, 3.5 kV; cylinder, 3.5 kV; capillary exit, 100 V; skimmer voltage, 10 V, hexapole offset, 2 V; hexapole RF, 700 Vpp; exit lens, –100 V and an injection low m/z ratio cut-off of 40 Th. Axial modulation is also applied to enhance ion resolution. Heterodimer ions were selected by broadband isolation mode and submitted to resonant excitation for collision-induced dissociations with the helium buffer gas. The isolation width of selected ions in MS/MS experiments was set to 4 Th. All chemical compounds were purchased from Sigma Aldrich Chemical (France).