

1 Introduction
In recent years there has been considerable interest in the use of proton ionisable crown compounds for the transport of metal cations between two aqueous solutions through an organic membrane (see [1], and references therein). Thus, compounds with pendant arms containing carboxylic or phenolic groups have been synthesised and used as carriers in transport experiments [2]. Lately, carboxylic or phenolic groups have been changed by hydroxamic acids, carbamoyl functions or phosphonic acid monomethyl ester, and new ligands have been synthesised to be used in a similar way [3].
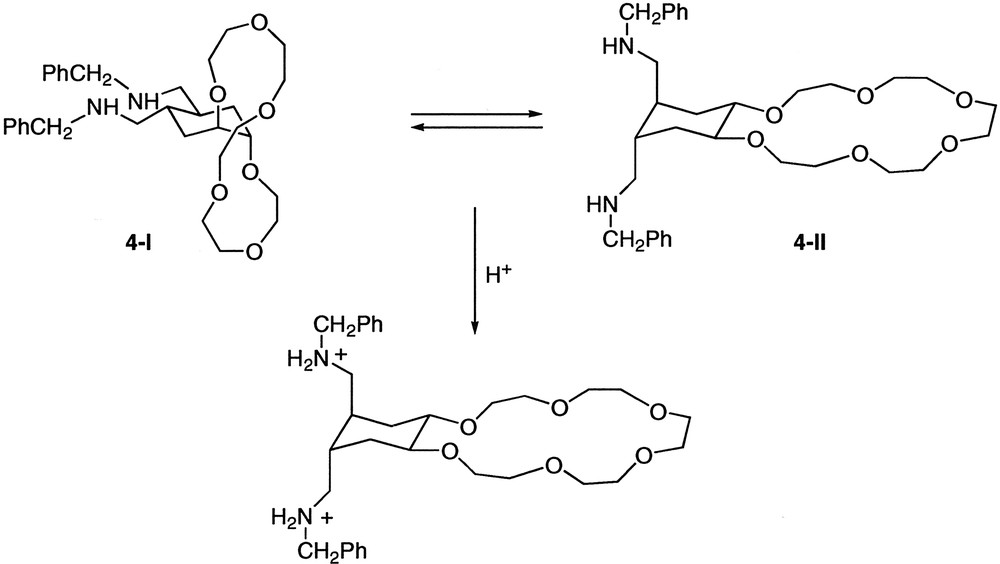
The pH-regulated ligands described until now present regulation due to electrostatic effects or the fact that transfer of the counter-anion from the aqueous phase into the organic medium is avoided in some cases. However, regulation produced by pH induced conformational changes has been studied less. For this reason, we have prepared some pH-dependent ligands using cyclohexane moieties as transmitters of conformational information. The rigidity experimented by a crown ether directly bound to a cyclohexyl moiety has been previously exploited by our research group in controlling the ability of ligands 1 and 2 to complex and transport cations [4] (Fig. 1 ). Thus, the positive allosteric cooperativity observed in these compounds widely increased the transport of cations through organic membranes. The allosteric behaviour observed in these ligands was due to the stereochemistry of the cyclohexane derivative, in which only one site at a time has a favourable conformation for binding. In the same way, the rigidity of the cyclohexyl system provides a relationship between the crown ether and the substituents on the cyclohexane ring. Thus, it has been reported that for trans-cyclohexyl crown ethers with a bulky substituent in 3-, conformations with the equatorial substituent are more stable than those with the axial disposition [5]. On the other hand, the use of pH dependent substituents on the cyclohexane allowed the control of complexation, by making changes in the pH of the medium [6]. Making use of all these principles we have prepared compounds 3 and 4 (Fig. 1) which should show different conformation of the cyclohexane system depending on the pH of the medium. Thus, the stereochemistry of compound 3 should make it able to complex cations only if the crown ether moiety presented a trans-diequatorial conformation (lone pair on the oxygen atoms directed to the centre of the cavity). In addition, the rigidity of the cyclohexyl moiety would fix the conformation in such a way that compound 3 should be able to form more stable complexes under basic conditions than in an acidic medium. The opposite behaviour should be expected for compound 4 (Fig. 2 ). In this case, an acidic medium should favour a trans-diequatorial conformation of the crown ether, due to the electrostatic repulsions between the two positively charged ammonium groups.
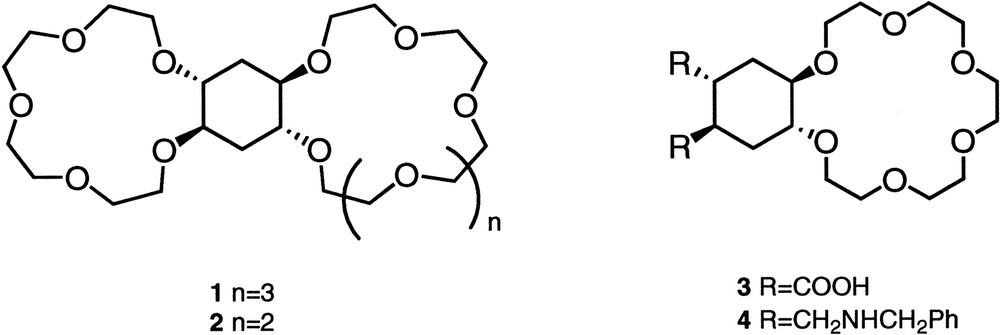
2 Discussion and results
2.1 Synthesis and conformational studies in solution
The synthesis of compound 3 and studies of its conformational equilibrium in solution have been previously described (potentiometric titrations carried out with compound 3 showed that this compound is monoprotonated at pH = 7.0) [7]. We would like to report here the preparation of compound 4 (Fig. 3 ) and its conformational behaviour in solution. Thus, compound 5 [8] was converted into the corresponding diamide 6 by reaction with benzylamine and trimethylaluminum in toluene [9]. For the subsequent reduction of 6 to give compound 4, several conditions and reducing agents were studied. When BH3 or NaBH4/CH3COOH were used, [10], the starting material was always recovered. On the other hand, the use of Red-Al® in toluene as reducing agent [11] led to the corresponding imine 7, but all attempts to reduce the imine to amine 4 by catalytic hydrogenation on Pd/C or with Rh/alumina were unsuccessful. Compound 4 could finally be obtained (84% isolated yield) by reaction of 6 with a large excess of LiAlH4 in refluxing THF for two days. All compounds have been characterized by 1H and 13C NMR spectroscopy and FABMS spectrometry. The solid-state structure of amide 6 could be determined by X-ray diffraction, confirming the trans-anti-trans stereochemistry of the cyclohexane ring.
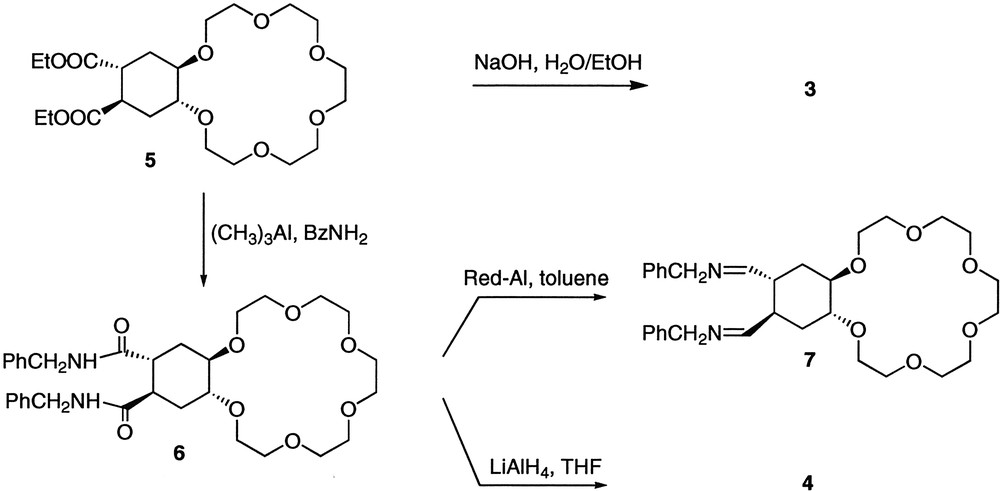
Conformational studies carried out with compound 4 demonstrated that a conformational equilibrium between both chair conformations exists in CDCl3. These studies were carried out using 1H NMR techniques and it was also observed that conformational changes are relatively slow (Fig. 4 ). Thus, compound 4 showed a broad signal at 1.65 ppm corresponding to Hax and Hb. Additionally Heq appeared like a broad singlet at 1.70 ppm and Ha was included with the –CH2-O- hydrogen atoms in the broad signal at 3.55–3.70 ppm. On the other hand, COSY experiments demonstrated that both Hb and Ha showed correlations with Hax and Heq in accordance with an equilibrium between similar amounts of both conformations.
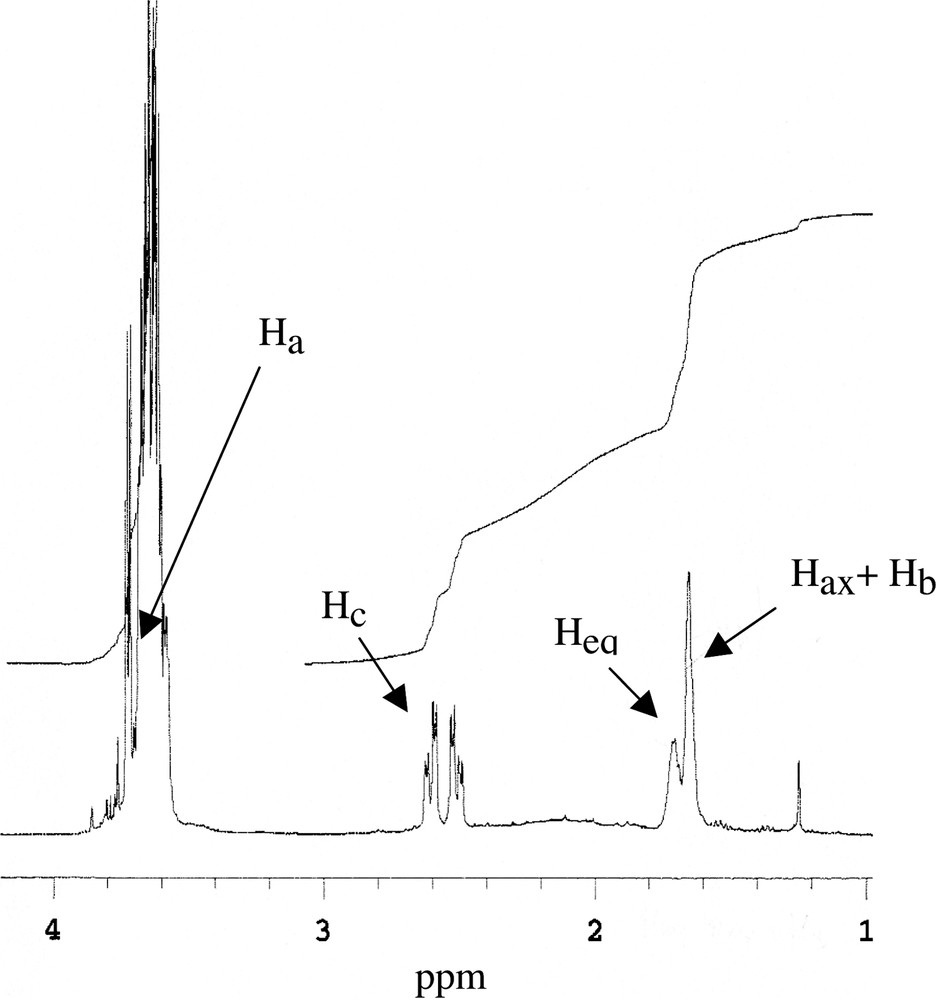
1H MNR of compound 4 in CDCl3.
COSY, NOE and NOESY experiments were carried out to confirm the conformational changes of compound 4 in an acidic medium. NOE experiments were not conclusive because in both conformations (4-I and 4-II) the molecule possesses a C2 symmetry axis which makes both sides of the cyclohexane ring equivalent. Coupling constants would give more information about the spatial disposition, but unfortunately slow exchange on the NMR time scale led to broad signals (even over a range of temperatures). The intensity of the cross peaks in COSY experiments turned out to be the most reliable proof of the preferred conformational arrangement (Fig. 5 ). These experiments showed that compound 4 was present in only one fixed conformation in acidic (CF3COOH) solutions of CDCl3. Under these conditions, the benzylic protons are downfield shifted (from 3.72 to 4.20 ppm), similarly Hb is shifted from 1.65 to 2.38 ppm and finally, Heq and Hax appeared now as two different signals at 1.85 and 1.65 ppm respectively. The main conformation in these conditions was that with the protonated amines in trans diaxial position. This result was the expected because in this conformation both charged groups are far away from one another. This affirmation agreed with the strong correlation observed between Hax and Ha and the weaker correlation also observed between Heq and Hb.
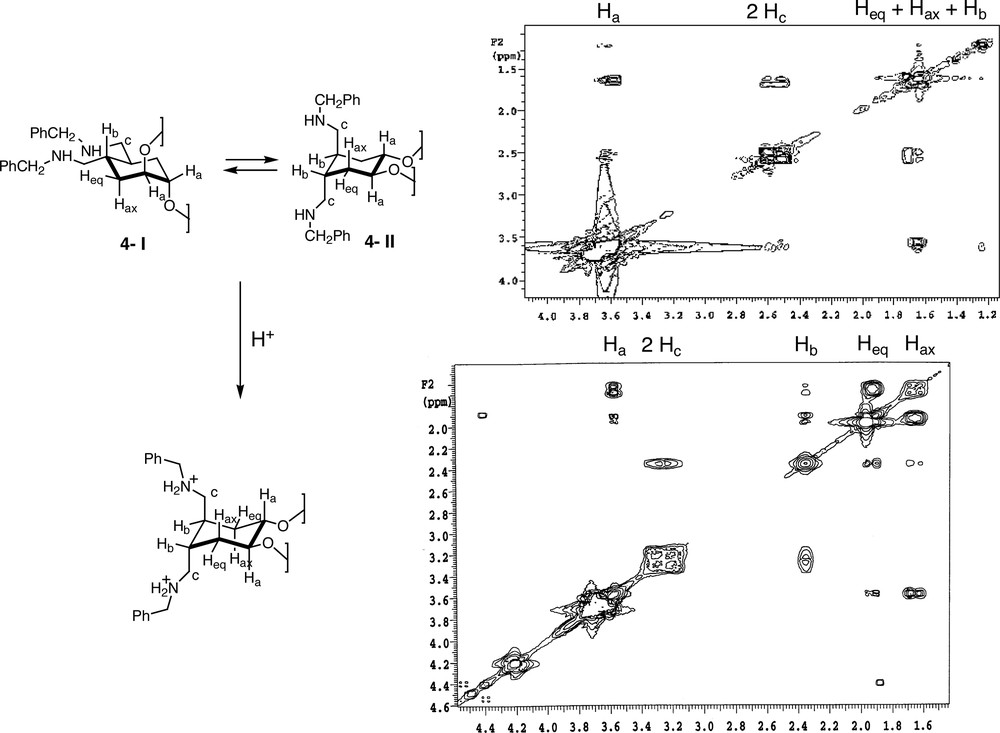
COSY experiments of ligand 4 in CDCl3; upper: the free amine; lower: the protonated compound.
Finally, 1H NMR experiments demonstrated that the presence of bases able to form hydrogen bonds (such as H2NC(CH2OH)3) stabilizes the conformation with the amino groups in trans diequatorial positions.
2.2 X-ray determination
The relative trans-anti-trans stereochemistry of compound 4 was definitively established by X-ray diffraction of the amide precursor, compound 6 (Fig. 6 ). Single crystals were obtained by slow diffusion of hexane into a CHCl3 solution. Table 1 summarises information concerning bond distances and angles. The molecular structure shows the cyclohexyl ring on a chair conformation with the benzylcarbamoyl groups in trans-diequatorial positions. There are not appreciable distortions in the cyclohexane ring, only a small increment in the values of the angles C(11)–C(13)–C(15) and C(16)–C(14)–C(12) (112.8° and 112.0°, respectively) was observed. On the other hand, separation from the normal values were observed for C(15)–C(17)–N(1) and C(16)–C(18)–N(2) angles (114.4° and 114.9° respectively). Finally, there is a clear distortion in the crown cavity; thus, the angle C(5)–C(6)–O(4) shows a value of 96° and at the same time, bond distances O(4)–C(6), C(6)–C(5) and O(4)–C(7) with values of 1.35, 1.59 and 1.46 Å, respectively, are longer than expected in this type of compounds. By contrast, in the opposite part of the crown angles C(1)–C(2)–O(2) and C(2)–O(2)–C(3) show values (124.4° and 119°, respectively) higher than those expected for sp3 atoms.

Ellipsoid plot of compound 6.
Selected bond distances and angles for ligand 6
| Bond angles/bound distances | ° / Å |
| O(4)–C(6)–C(5) | 96(2) |
| C(6)–O(4)–C(7) | 114(2) |
| O(4)–C(7)–C(8) | 112.1(12) |
| C(1)–C(2)–O(2) | 124.4(14) |
| C(2)–O(2)–C(3) | 119.0(2) |
| C(15)–C(17)–N(1) | 114.4(8) |
| C(15)–C(17)–O(7) | 121.2(7) |
| O(7)–C(17)–N(1) | 124.2(9) |
| C(16)–C(18)–N(2) | 114.9(8) |
| C(16)–C(18)–O(8) | 123.2(7) |
| O(8)–C(18)–N(2) | 121.8(10) |
| O(4)–C(6) | 1.35(2) |
| C(6)–C(5) | 1.59(2) |
| O(4)–C(7) | 1.46(2) |
2.3 Transport experiments
The study of compounds 3 and 4 as pH-dependent switches was carried out using transport experiments. Experiments were carried out in a U-tube [12] where two aqueous phases were separated through an organic membrane (CHCl3). One of the aqueous phases, the source phase, was a solution of the salt studied (KPic, KCl, SrPic2 and SrCl2) and the other, the receiving phase, was deionised water. Experiments were carried out with ligands 3, 4 and control ligand 5. Ligand 5 bears two etoxycarbonyl groups as substituents in the cyclohexane moiety, and for this reason the conformation of the crown cavity should not be modified by pH. So transport experiments with this ligand can be used to evaluate the influence of the pH-dependent groups of ligands 3 and 4 on their transport abilities. Two different techniques (UV-vis and atomic absorption) were used to determine the amount of salt transported. UV-vis spectra allowed us to evaluate the amount of picrate anion transported from the source phase to the receiving phase, whereas atomic absorption determinations gave us the total amount of cation in the receiving phase. Both values can be equal or different depending on pH conditions during the experiment. The amount of salt in the receiving phase has been expressed using the parameter transport efficiency (E). This parameter was defined as E = [(μmol cation transported / ml receiving phase)/mmol carrier] [13]. The results obtained in the different experiments, after 24 h of transport, are shown in Table 2.
Transport efficiency (after 24 hours) with ligands 3, 4 and 5
| Ligand | pH source phase | pH receiving phase | KPic (UV) | K+ (AA) | SrPic2 (UV) | Sr2+ (AA) |
| 5 | 7.0 | 7.0 | 160.7 | 159.8 | 82.8 | 79.5 |
| 3 | 7.0 | 7.0 | 38.0 | 38.6 | 18.1 | 20.1 |
| 3 | 11.0a | 7.0 | 177.1 | 203.1 | 129.9 | 150.6 |
| 4 | 7.0 | 7.0 | 55.3 | 55.1 | 55.0 | 54.8 |
| 4 | 7.0 | 11.0a | 48.9 | 16.8 | 47.3 | 28.2 |
| 4 | 11.0a | 7.0 | 42.6 | 70.1 | 35.5 | 51.7 |
| 4 | 11.0a | 2.0b | 30.8 | 85.4 | 31.7 | 65.4 |
a pH conditions were regulated with Me4NOH.
b pH conditions were regulated with HCl.
2.3.1 Experiments with ligand 3
Ligand 3 presents two different moieties able to interact with cations: (a) the crown cavity; and (b) the carboxylate groups generated from the carboxylic acids under a basic medium. However, transport of picrate anion can only be carried out forming an ionic pair with the cation complexed inside the crown cavity. Thus the evaluation of the concentration of picrate anion at the receiving phase gives us information about the cation transported only inside the crown ether cavity. By contrast, the atomic absorption values indicate the total amount of transported cation, independently of the part of the molecule involved in the transport.
Taking into account the former ideas, two types of experiments were carried out. Firstly, pH = 7.0 was used in both the source phase and the receiving phase, and transport efficiencies for potassium after 24 h were found to be 38.0 and 38.6 respectively, when UV-vis and atomic absorption were use as detection methods. These results indicated that the concentration of picrate in the receiving phase was almost equal to the total concentration of potassium. Thus, ligand 3 transports the cation only inside the crown cavity even though this compound should be partially dissociated in water, forming the monopotassium salt at the source interphase [7]. In any case, the value of 38.0 is lower than that observed in control experiments with ligand 5. This difference could be due to the formation of an intramolecular hydrogen bound in compound 3 that would shift the conformational equilibrium toward the less suitable conformation for complexation, that is, a trans-diequatorial disposition of the carboxylic groups and a trans-diaxial conformation of the crown ether moiety.
When pH values of 11.0 (Me4NOH) at the source phase and 7.0 at the receiving phase were used, much higher transport efficiencies for potassium were observed. UV determinations of picrate showed a high transport inside the crown cavity (E = 177.1). However, the potassium cation was also transported as counterion due to the negative charge of the carboxylate groups (the efficiency determined using atomic absorption was 203.1). The observed results seem to agree with a cotransport of potassium due to the charge of the ligand. The higher efficiency of the transport inside the crown cavity observed under these conditions could be related to the presence in the membrane of a conformation with the carboxylate groups in a trans-diaxial conformation and the crown cavity in trans-diequatorial. Once the carrier arrives to the receiving phase interphase, at pH = 7, the dicarboxylate forms the monopotassium salt, [7] favouring a trans-diaxial conformation of the crown ether which would release the cation (Fig. 7 ).
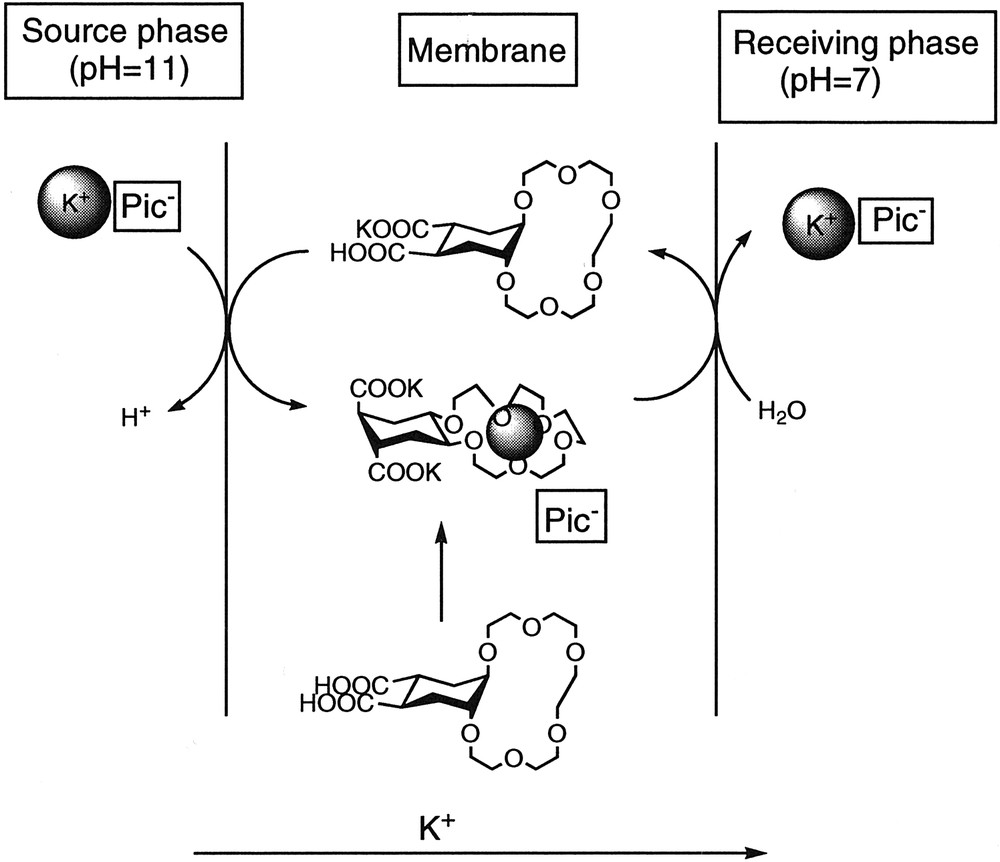
Transport experiment with ligand 3 at different pH.
Similar tendencies were found in transport experiments with SrPic2 (see Table 2). As would be expected due to the results obtained with the control ligand 5, lower E values were observed in both experiments. These results can be explained on basis of the smaller size of this cation and the double positive charge in it.
2.3.2 Experiments with ligand 4
First of all the experiment was carried out keeping pH = 7.0 in both the source phase and the receiving phase. Under these conditions, at the source phase interphase, the ligand should be monoprotonated (log K = 8.32) the corresponding counterion being picrate anion. Anyway, the picrate anion remains in the membrane, but is not transferred into the receiving phase, as similar E values for potassium were obtained by UV-vis (55.3) and atomic absorption (55.1). Thus, the concentration of picrate determined in the receiving phase should be related to the KPic transported inside the macrocyclic cavity. These results are in accordance with the ones reported by Ikeda et al. [14]. In this experiment, the transport efficiency was lower than with the control ligand 5 (160.7 by UV). This behaviour could be due to the formation of a hydrogen bond between the free NH and the protonated one. This interaction would modify the conformational equilibrium, shifting it toward the trans-diaxial conformation of the crown ether, which makes complexation at the source phase less efficient, thus decreasing the transport of the cation.
When the pH was 7.0 at the source phase and 11.0 at the receiving phase, ligand 4 should be monoprotonated at the source phase interphase, but showing both amino groups free at the receiving phase. Under these conditions, concentration of picrate at the receiving phase was not directly related to the cation transported, because the picrate which was also transported as counterion in the monoprotonated ligand, is now released when arriving to the receiving phase interphase. Thus, the concentration of potassium cation evaluated by atomic absorption gives rise to an efficiency of 16.8, whereas the UV determination gives an E value of 48.9. One explanation of the lower transport efficiency observed as compared with the neutral conditions (16.8 versus 55.1) can be found in the absence of positive charges in the ligand at the receiving phase. This would make complexation stronger under basic conditions, giving rise to a lower release of the transported cation to the receiving phase.
When we fixed a pH = 11 at the source phase and pH = 7.0 at the receiving phase, the observed transport efficiency value for potassium (70.1 by AA) was higher than in the former cases. This result would be in agreement with a transport regulated by Coulombic repulsions that would make complexation at the source phase (free amino groups) and also releasing at the receiving phase (monoprotonated ligand) easier under these pH conditions. In this case, we found a lower E value when using the UV technique (42.6). We think that this difference between the amount of potassium and the amount of picrate at the receiving phase is due to a transport of the picrate in the reverse direction (from the receiving to the source phase), as counterion of the monoprotonated ligand. Finally, when pH = 11 and pH = 2 (HCl) were used at the source and receiving phases respectively, a similar behaviour was observed. However, these results are not too reliable due to the generation of picric acid, which is able to be diffused into the membrane.
When SrPic2 was used in similar experiments the results were those reflected in Table 2 and they can be explained on the basis of the former reasons. Finally, experiments carried out using chloride compounds (KCl and SrCl2) gave rise to transport efficiencies lower than 1 in all the conditions studied. This is very likely due to the lower lipophylicity of the chloride anion.
3 Conclusions
Transport of KPic and SrPic2 through a liquid membrane using pH-dependent carriers 3 and 4 is influenced by the pH of the source and receiving phases. The studies carried out with ligand 4 have demonstrated that transport is really determined by an electrostatic reason. On the other hand, in the case of ligand 3 the increment in transport observed when there is a basic pH at the source phase seems to be related to the influence of pH on the conformational equilibrium.
4 Experimental section
4.1 General methods
All commercially available reagents were used without further purification. Toluene and THF were distilled from Na and Na/benzophenone prior to use. Melting points are uncorrected. NMR spectra were recorded on either Bruker AC250 or Varian Unity 300/400 spectrometer. Chemical shifts in ppm are referenced downfield from TMS. Mass spectra were taken on a VG-AUTOESPEC spectrometer. UV determinations were carried out in a spectrophotometer Shimazdu UV-2101 PC. Atomic Absorption was measured in an Atomic Absorption Spectrophotometer Perkin-Elmer 2380.
4.1.1 Compound 6
1.14 ml (2.27 mmol) of a 2 M THF solution of trimethylaluminium was poured into 10 ml of anhydrous toluene. 248 μl (2.27 mmol) of benzylamine were added to this solution at 0 °C. The mixture was kept at –15 °C for 2 h and then it was allowed to rise to room temperature. 350 mg (0.757 mmol) of 5 in 10 ml of toluene were added to the former solution and the reaction was kept under reflux for 16 h. The reaction was allowed to reach room temperature and then 10 ml of water were added. The organic solvent was dried with anhydrous Na2SO4 and evaporated to give a solid. The solid was washed with hexane and petroleum ether to give 6 as a white solid (70%). m.p. 110–112 °C. 1H NMR (250 MHz, CDCl3): δ 7.26–7.13 (m, 5H), 6.10 (bs, 1H), 4.35 (dd, J = 14.8 and 6.1 Hz, 1H), 4.14 (dd, J = 14.8 and 5.4 Hz, 1H), 3.77 (bs, 1H), 3.70–3.52 (m, 10H), 2.73–2.68 (m, 1H), 2.05–1.93 (m, 1H), 1.79 (bd, J = 14 Hz, 1H); 13C NMR (62.5 MHz, CDCl3): δ 174.8, 138.3, 128.5, 127.6, 127.2, 73.8, 71.3, 70.6, 68.2, 43.1, 41.3, 28.5; HRMS (IE)(M+) calc. for C32H44O8N2 m/z 584.3099; found 584.3099.
4.1.2 Compound 7
326 mg (6.8 mmol) of Red-Al® (65%) were added to a previously cooled (0 °C) solution of 127 mg (0.217 mmol) of compound 6 in 15 ml of dry toluene. The suspension was heated during 2 h and then EtOH and NaOH (10%) were added. The organic phase was evaporated and the solid purified by column chromatography (alumina, CH2Cl2 /MeOH 95:5) to give 7 (75%, mixture of isomers) as a white solid. 1H NMR (250 MHz, CDCl3): δ 7.45–7.17 (m, 12 H), 5.38 (d, J = 15 Hz, 1 H), 5.02 (d, J = 15 Hz, 1 H), 4.96 (d, J = 16 Hz, 1 H), 4.71 (d, J = 16 Hz, 1 H), 3.80–3.42 (m, 18 H), 3.41-3.02 (m, 4 H), 2.57 (t, J = 12 Hz, 1 H), 2.40–2.18 (m, 2 H), 1.92–1.73 (m, 2 H), 1.54 (t, J = 13 Hz, 1 H); 13C NMR (62.5 MHz, CDCl3): δ 168.3, 136.8, 133.4, 129.0, 128.8, 128.5, 127.4, 126.3, 74.2, 73.9, 71.2, 71.1, 70.6, 68.4, 68.1, 55.2, 51.3, 45.9, 43.5, 36.4, 27.9, 27.2; HRMS (FAB+): calcd. for C32H45N2O6 (MH+) m/z 553.3278; found 553.3272.
4.1.3 Compound 4
300 mg (6.8 mmol) of LiAlH4 were added to a solution of 200 mg of 6 in 20 ml of dry THF. The suspension was heated at reflux during 20 h and then 2 ml of water were added. The solution was treated with HCl to reach pH < 2 and the reaction was stirred for 30 min. Subsequently the solution was turned basic with NaOH and extracted with EtOAc. The organic phase was dried with anhydrous Na2SO4 and evaporated to give 4 as an yellow oil (86%). 1H NMR (250 MHz, CDCl3): δ 7.26 (m, 10H), 3.72 (s, 4H), 3.55-3.70 (m, 22H), 2.59 (dd, J = 11.6 and 5 Hz, 2H), 2.51 (dd, J = 11.6 and 5 Hz, 2H), 1.68 (m, 2H), 1.65 (m, 4H); 13C NMR (62.5 MHz, CDCl3): δ 139.2, 128.3, 128.1, 127.0, 76.2, 70.6, 68.3, 53.5, 52.6, 35.0, 29.8; HRMS (FAB+) (M+) calc. for C32H48O6N2 m/z 556.3515; found 556.3515. Combustion analysis calc. for C32H48O6N2: 69.06% C, 8.63% H, 5.03% N; found 69.10 % C, 8.70% H, 5.01% N.
4.2 Transport experiments
Compounds 3 and 5 were prepared as described in [7] and [8]. Transport experiments were made in a U-tube cell at 25 °C. A solution 0.003 M of the ligand in free-ethanol chloroform (12 ml) was placed in the bottom of the cell, and two portions of aqueous solution (2 ml) were carefully added each of them on the top of one of the lateral arms. The source phase was a 0.01 M aqueous solution of the corresponding salt and the receiving phase was deionised water. When modification of pH was carried out, 0.01 M HCl and 0.15 M tetramethylamonium hydroxide were used. Both surface areas were 1.75 cm2. The organic phase was magnetically stirred at 500 rpm. The concentration of the picrate anion in the receiving phase after 24 h was obtained by calculation based on the absorption at 354.0 nm for KPic and 356 nm for SrPic2 in the UV spectrum. Direct evaluation of cation concentration was carried out by atomic absorption. Each experiment was repeated at least three times, and the results reported are the average of the three determinations. The standard deviation from the mean value among the data was lower than 5% and diffusion was determined through blank experiments.
4.3 X-ray determination
Intensity measurements were made on a Siemens PA diffractometer using a colourless single crystal of dimensions 0.20 × 0.25 × 0.26 mm. Graphite-monochomated Mo Kα radiation (λ = 0.71073 Å). Data collection was carried out at room temperature. Three reference reflections were measured every two hours as an intensity and orientation check and no significant fluctuation was noticed during the collection of the data. The Lorentz-polarization correction was made. The crystal structure was solved by direct methods using SHELXS system [15] and refined by full matrix least-squares techniques [16] on F2. The hydrogen atoms were geometrically generated with SHELXL-93 program with fixed isotropic displacements parameters. Fig. 4 was made with ORTEP [17] with thermal ellipsoids set at a 50% probability level. It is possible to ask for additional material to this paper, referring to deposit CCDC 196475 at the Cambridge Crystallographic Data Centre, CCDC, 12 Union Road, Cambridge CB2 1EZ, United Kingdom.
Acknowledgements
We acknowledge the support of the EC, project F14WCT960022 and DGITYC, project PB98-1429.
Vous devez vous connecter pour continuer.
S'authentifier