

Cyclodextrins (CDs) are natural polyhydroxylated molecules, made out of d-glucopyranose units, with a unique hollow-truncated-cone geometry [1]. They function as host molecules, forming inclusion complexes with a wide variety of hydrophobic guest compounds in aqueous solution [2]. Per-O-methylated CDs have attracted considerable attention due to their solubility both in water [3] and in organic solvents [4]. On the other hand, inclusion complexes of methylated CDs are usually more stable than the corresponding complexes of unmodified CDs [3]. For these reasons, the controlled chemical synthesis of modified methylated CDs having a few specifically located hydroxyl groups available for the preparation of more elaborate molecular systems represents a true challenge for synthetic chemists [5]. This is especially true for the widely used β-CD derivatives. Logically enough, available [6] methods proceed through a temporary regioselective protection of peculiar hydroxyl groups of the native CD, followed by O-methylation and final removal of the protective groups to unmask the required hydroxyl functions.
We recently introduced [7] a conceptually new way to directly get such compounds. It is based on the efficient selective de-O-alkylation of a fully alkylated α or β-CD, using commercially available diisobutylaluminum hydride (DIBAL) as a regioselective chemical ‘scalpel' [8]. As an original example [7], DIBAL is achieving a remarkable AD type regioselective bis-de-O-benzylation on the primary upper rim of either α or β perbenzylated CDs. The resulting diols, directly obtained through such an unprecedented approach, are well suited for further transformations [9]. It was then discovered [10] that permethylated α or β-CDs presented a strikingly different behaviour. For example, when commercially available permethylated β-CD 1 was reacted with DIBAL (7 equiv) in toluene at 50 °C for 3 h (Fig. 1 ), a remarkably direct regioselective bis-de-O-methylation on the secondary rim was observed, giving the easy isolable 2A, 3B diol 2 in 55% yield. An isolated by-product (20% yield) was the 6A alcohol 3. We would like now to disclose a practical and unprecedented regioselective tetra-O-demethylation of permethylated β-CD 1, using a large excess of DIBAL as a chemical ‘scalpel'.

Reagents and conditions: DIBAL (7 equiv), toluene, 50 °C, 3 h.
When permethylated β-CD 1 was treated with 50 equiv of DIBAL in toluene at room temperature for 3 h (Fig. 2 ), we observed the formation of four products, which have easily been separated by conventional silica gel column chromatography, and easily identified as mono-, di-, tri- and tetra-O-demethylated derivatives by FAB-MS, with yields of 1, 8, 17, and 51%, respectively. Mono- and di-O-demethylated compounds were identified as the previously prepared compounds 3 and 2. NMR study of triol revealed a mixture of two isomers, which was not analysed further at this stage. A most remarkable outcome of this reaction was the easy isolation, in about 50% yield, as a major product, of a tetrol wherein four methyl groups have selectively been stripped off. This tetrol 4 was a single isomer, as confirmed by acetylation to the tetracetate 5: 1H NMR spectrum showed four acetyl groups at δ 2.19, 2.19, 2.21 and 2.23 ppm; 13C NMR spectrum showed four methyl and carbonyl groups of acetate at δ 21.15, 21.24, 21.42, 21.46 and 169.85, 169.91, 170.39, 170.52 ppm, respectively. The structural assignment of tetrol 4 was next achieved through a careful combined analysis [11] of homonuclear 2D NMR spectra and their semi-selective version: COSY, relayed COSY (simple, double and triple relay) and off-resonance ROESY (effective angle = 54.7°), as well as heteronuclear 2D NMR spectra: HSQC and HMBC. The overlapping signals of the initial 1H NMR spectra were first separated through relayed COSY into sets of proton resonances belonging to the same glucopyranose unit. It was found that the four hydroxyl groups were carried by four different glucopyranose units, two of them being C2–OH, and the two others C3–OH. The two direct glycosidic linkages A–B and D–E have been demonstrated through correlations from off-resonance ROESY spectra, which have been further confirmed by the correlations C1A–H4B and C1D–H4E of HMBC spectra. The relative positioning of sugar rings was obtained without ambiguity by the following correlations on the HMBC spectra: C4D–H1C, H4D–C1C, C4C–H1B, and H4C–C1B. The NMR (500 MHz) assignments of tetrol 4 are shown in Table 1.
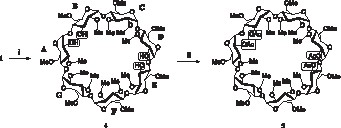
Reagents and conditions : (i) DIBAL (50 equiv), toluene, RT, 3 h; (ii) Ac2O, pyridine, RT, 24 h.
13C and 1H NMR (500 MHz) chemical shiftsa for tetrol 4 in DMSO-d6 at 25 °C.
| A | B | C | D | E | F | G | |
| C1 | 105.7 | 103.0 | 102.4 | 105.6 | 103.0 | 102.4 | 102.5 |
| C2 | 77.24 | 85.48 | 85.80 | 77.05 | 85.48 | 85.79 | 85.79 |
| C3 | 87.54 | 75.58 | 85.73 | 87.41 | 75.56 | 85.73 | 85.79 |
| C4 | 84.11 | 85.54 | 84.74 | 84.30 | 85.56 | 84.86 | 84.23 |
| C5 | 75.16 | 75.55 | 74.82 | 75.22 | 75.52 | 74.80 | 74.61 |
| C6 | — | — | — | 75.54 | — | — | — |
| H1 | 5.368 | 5.616 | 5.620b | 5.373 | 5.604 | 5.616b | 5.622 |
| H2 | 3.953 | 3.614 | 3.648 | 3.952 | 3.611 | 3.645 | 3.648 |
| OH2 | 6.251 | 6.234 | |||||
| H3 | 3.854 | 4.311 | 3.889 | 3.851 | 4.305 | 3.883 | 3.901 |
| OH3 | 6.500 | 6.470 | |||||
| H4 | 3.960 | 3.974 | 4.022 | 3.982 | 3.985 | 4.020 | 4.010 |
| H5 | 4.357 | 4.327 | 4.271 | 4.345 | 4.327 | 4.273 | 4.298 |
| H6 | — | — | — | 4.289 | — | — | — |
| H6' | — | — | — | 4.105 | — | — | — |
a 1H and 13C methoxy signals are observed through HSQC spectra but remain not assigned.
b These values could be exchanged each other.
In contrast with the more hindered perbenzylated CD derivatives, the two contiguous and closely located methoxy groups at C-2 and C-3 are kinetically selected by DIBAL. In the presence of a large excess of DIBAL, a second and opposite corresponding pair is stripped off, the origin of the selectivity being probably steric. This corresponds in a way to the AD type primary rim bis-de-O-benzylation [7] of fully benzylated CDs.
In conclusion, this piece of work demonstrates that the concept of DIBAL-mediated selective defunctionalisation compares well with the more classical route based on selective functionalisation. It provides, in a single chemical operation, modified CDs having a substitution pattern that is often different from the one attainable by the classical approach.
1 Experimental
1.1 Protocol for the practical preparation of the tetrol 4
To a stirred solution of permethylated CD 1 (1.0 g, 0.7 mmol) in anhydrous toluene (20 ml) was added 23.4 ml (50 equiv, 1.5 M in toluene) of DIBAL at room temperature under argon. The reaction mixture was stirred at this temperature for 3 h. The solution was cooled to 0 °C, quenched with aqueous HCl (1 M), and the mixture was stirred vigorously at room temperature for 30 min. The toluene phase was collected, and the aqueous phase was extracted by ethyl acetate (3 × 50 ml). The combined organic phases were washed with brine, dried over MgSO4, and the solvent was removed in vacuo. Purification of the crude product by silica gel chromatography, eluting with 96:4 dichloromethane/methanol, afforded 492 mg (51%) of 4 as colourless foam. [α]D20 +152 (c 1.3, CHCl3); 1H and 13C NMR (DMSO-d6): see Table 1. FAB-MS: m/z 1395.6 (100%, M+Na+), CI–MS: m/z 1390.7 (100%, M+NH4+). Anal. calcd for C59H104O352 H2O (1409.48): C, 50.27; H, 7.72. Found: C, 50.28; H, 7.77.
Acknowledgements
YC thanks the French Ministry of Research and Technology for post-doctoral fellowship, and XL the China Scholarship Council for financial support. We thank Cyclolab (Hungary) for a generous supply of starting material 1, and Drs H. Desvaux and P. Berthault for technical assistance and valuable discussions.