

1 Introduction
Homogeneous catalysis plays a major role in modern synthetic chemistry. The catalyst usually features a metal and a ligand and its efficiency is dependent on both the nature of the metal and the structure of the ligand. Among the latter, phosphine ligands have a place of choice [1]. Their steric and electronic effects often play key roles in determining organometallic reactivity trends and catalytic properties [2]. Despite their importance, there is no easy and general way to prepare phosphines and chemists usually restrict themselves to the limited number of commercially available ones. In particular, few methods have been reported for the synthesis of electron-rich alkylarylphosphines. Important ways include reactions between haloarylphosphines and alkyl-magnesium or lithium derivatives and nucleophilic substitution reactions of alkyl halides [3]. However, these methods are often troubled by a large number of by-products and by the air sensitivity of phosphines that leads to modest yields. To circumvent these problems, new methodologies using phosphine boranes derivatives (phospha-anion, chlorophosphines…) have been depicted [4–6]. Nevertheless, both from economic and environmental points of view, developing new, versatile and selective synthetic routes to electron-rich alkylarylphosphines from inexpensive feedstock is of great interest. An environmentally benign one-pot, atom-efficient synthesis of phosphines from ubiquitous alkenes is the hydrophosphination reaction, a well-known industrial process. This reaction involves phosphines such as PH3, primary and secondary derivatives and is mainly carried out in the presence of radical initiators [7–10]. A few recent examples reported the use of a catalytic activation [11–13]. Even if some of these reactions can be efficiently carried out, this methodology still needs to be improved with regard to generality and efficiency. Recently we [14–16] and others [4–6] have shown that phosphine borane complexes can advantageously replace ‘free’ phosphines in synthesis. Temporary protection of the phosphorus with BH3 [17–23] allows convenient handling, purification and storage of phosphines. Beside its protective function, the borane group also activates the hydrogen carried by the phosphorus atom [17]. Furthermore, phosphine borane complexes can be used directly as pro-ligand in catalysis [24]. We recently showed that phosphine borane complexes could be advantageously used for the synthesis of vinylphosphine derivatives by hydrophosphination of unactivated alkynes [25]. Expanding this methodology to unactivated alkenes would provide an easy access to alkyl-substituted phosphines. To our knowledge, there is only one previous article dealing with the addition of secondary phosphine boranes onto unactivated alkenes in the literature [26] and the product yields remained low (7 to 35%). The present contribution describes a facile gram-scale synthesis of P-alkyl substituted phosphine derivatives using this hydrophosphination methodology (for a preliminary communication, see [27]).
2 Experimental
2.1 General comments
All glassware was flamed before use. Toluene was purified by refluxing and distillation over sodium. All alkenes were distilled before use. Phosphine borane precursors 1, 8, 11, on the one hand, and 3, on the other hand, were respectively prepared according to literature procedures [26] and [28].
The NMR spectra (1H, 13C and 31P) were recorded either on a Bruker AC 300P or on a Bruker AC 400 spectrometer. HRMS (High-Resolution Mass Spectrometry) experiments were performed on a Varian MAT 311 instrument using electron impact ionisation technique. The IR spectra were recorded on a Perkin-Elmer 16PC FT–IR spectrometer. The microwave device used is a Synthélabo Synthewave 402, with a maximum power of 300 W. The term RT in the text means a reaction carried out in a regulated oil bath, T = 25 °C, to avoid reproducibility problems.
2.2 General procedures
2.2.1 Method A: general procedure for microwave reactions
The phosphine borane (1 equiv) and a slight excess of alkene were placed in a quartz reaction vessel. The reactor was submitted to microwave irradiation for a time t, at the temperature T, and let to cool down to room temperature. The crude product was then purified by silica-gel column chromatography.
2.2.2 Method B: general procedure for oil-bath and room temperature reactions
Phosphine borane (1 equiv), alkene (1.2 equiv) and toluene were placed in a flamed nitrogen flushed Schlenk. The reaction was run for a given time t either at the temperature T or at RT. Reaction was followed by TLC until completion. The crude product was then purified by silica-gel column chromatography.
2.3 Experimental data
2.3.1 Experimental procedure for 2
Method A: diphenylphosphine borane 1 (100 mg, 0.5 mmol), oct-1-ene (392 μl, 2.5 mmol), t = 30 min, T = 50 °C. Yield: 68%. Method B: diphenylphosphine borane 1 (1 g, 5 mmol), oct-1-ene (950 μl, 6 mmol), toluene (15 ml), t = 48h, T = RT. Yield: 69%. The crude product was purified by silica-gel column chromatography with toluene as eluent (Rf = 0.7). Phosphine borane 2 was obtained as a colourless oil. 31P NMR (162 MHz, CDCl3): δ 16.1; 1H NMR (400 MHz, CDCl3): δ 7.67 (m, 4H), 7.44 (m, 6H), 2.19 (m, 2H), 1.51 (m, 2H), 1.37 (m, 2H), 1.25 (m, 8H), 1.00 (q, 1JHB = 98 Hz, 3H), 0.86 (t, 3JHH = 6.9 Hz, 3H); 13C NMR (101 MHz, CDCl3): δ 132.5 (d, 2JCP = 8.9 Hz), 131.5 (d, 4JCP = 2.3 Hz), 130.1 (d, 1JCP = 54.7 Hz), 129.2 (d, 3JCP = 9.8 Hz), 32.2 (s), 31.6 (d, 3JCP = 14.0 Hz), 29.5 (s), 29.4 (s), 26.0 (d, 1JCP = 37.0 Hz), 23.4 (s), 23.0 (s), 14.5 (s); 11B NMR (128 MHz, CDCl3): δ –36.7; HRMS calcd for C20H27P ([M–BH3]+•) 298.1850, found 298.1858.
2.3.2 Experimental procedure for 4
Method A: diphenylphosphine borane 1 (100 mg, 0.5 mmol), hex-1-ene (310 μl, 2.5 mmol), t = 30 min, T = 50 °C. Yield: 70%. Method B: diphenylphosphine borane 1 (50 mg, 0.25 mmol), hex-1-ene (38 μl, 0.3 mmol), toluene (600 μl), t = 72h, T = r.t. Yield: 65%. The crude product was purified by silica-gel column chromatography with toluene as eluent (Rf = 0.7). Phosphine borane 4 was obtained as a colourless oil. 31P NMR (121 MHz, CDCl3): δ 16.5; 1H NMR (300 MHz, CDCl3): δ 7.60 (m, 4H), 7.35 (m, 6H), 2.10 (m, 2H), 1.40 (m, 2H), 1.30 (m, 2H), 1.15 (m, 4H), 1.00 (q, 1JHB = 102 Hz, 3H), 0.75 (t, 3JHH = 6.8 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 132.6 (d, 2JCP = 9.7 Hz), 131.6 (d, 4JCP = 2.5 Hz), 131.1 (d, 1JCP = 55.0 Hz), 128.4 (d, 3JCP = 9.7 Hz), 28.2 (d, 1JCP = 32.7 Hz), 25.4 (s), 25.2 (d, 2JCP = 2.3 Hz), 23.4 (s), 18.2 (s), 12.4 (s); 11B NMR (128 MHz, CDCl3): δ –36.7; HRMS calcd for C18H23P ([M–BH3]+•) 270.1537, found 270.1533.
2.3.3 Experimental procedure for 5
Method A: diphenylphosphine borane 1 (100 mg, 0.5 mmol), 2,3,3-trimethylbutene (176 μl, 2.5 mmol), t = 30 min, T = 50 °C. The crude product was purified by silica-gel column chromatography with dichloromethane as eluent (Rf = 0.75). Phosphine borane 5 was obtained as a colourless oil in 64% yield. 31P NMR (121 MHz, CDCl3): δd 16.4; 1H NMR (300 MHz, CDCl3): δ 7.60 (m, 4H), 7.40 (m, 6H), 1.95 (t, 3JHH = 7.7 Hz, 2H), 1.70 (m, 1H), 1.00 (q, 1JHB = 95 Hz, 3H), 0.85 (s, 9H), 0.80 (t, 3JHH = 6.7 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 133.7 (d, 2JCP = 9.8 Hz), 131.4 (d, 4JCP = 2.3 Hz), 131.2 (d, 1JCP = 54.0 Hz), 129.1 (d, 3JCP = 9.7 Hz), 38.7 (d, 2JCP = 3.7 Hz), 34.2 (s), 29.0 (d, 1JCP = 34.9 Hz), 27.3 (s), 16.6 (d, 3JCP = 1.0 Hz); 11B NMR (128 MHz, CDCl3): δ –35.1; HRMS calcd for C19H25P ([M–BH3]+•) 284.1694, found 284.1689.
2.3.4 Experimental procedure for 6
Method A: diphenylphosphine borane 1 (30 mg, 0.15 mmol), allyltrimethylsilane (119 μl, 0.75 mmol), t = 25 min, T = 50 °C. Yield: 60%. Method B: diphenylphosphine borane 1 (80 mg, 0.4 mmol), allyltrimethylsilane (77 μL, 0.48 mmol), toluene (700 μL), t = 65h, T =RT. Yield: 62%. The crude product was purified by silica-gel column chromatography with toluene as eluent (Rf = 0.8). Phosphine borane 6 was obtained as a white powder (mp = 54 °C). 31P NMR (162 MHz, CDCl3): δ 15.3; 1H NMR (400 MHz, CDCl3): δ 7.66 (m, 4H), 7.44 (m, 6H), 2.24 (m, 2H), 1.55 (m, 2H), 1.00 (q, 1JHB = 98 Hz, 3H), 0.60 (m, 2H), –0.05 (s, 9H); 13C NMR (101 MHz, CDCl3): δ 132.5 (d, 2JCP = 9.0 Hz), 131.4 (d, 4JCP = 2.3 Hz), 130.2 (d, 1JCP = 54.5 Hz), 129.2 (d, 3JCP = 9.8 Hz), 29.8 (d, 1JCP = 34.9 Hz), 19.3 (d, 3JCP = 11.9 Hz), 18.2 (s), –1.2 (s); 11B NMR (128 MHz, CDCl3): δ –37.3. HRMS calcd for C18H25SiP ([M – BH3]+•) 300.1463, found 300.1480.
2.3.5 Experimental procedure for 7
Method A: dicyclohexylphosphine borane 3 (90 mg, 0.42 mmol), oct-1-ene (330 μl, 2.10 mmol), t = 50 min, T = 80 °C. The crude product was then purified by silica-gel column chromatography with toluene/pentane (1:1) as eluent (Rf = 0.45). Phosphine borane 7 was obtained as a colourless oil in 58% yield. 31P NMR (162 MHz, CDCl3): δ 24.2; 1H NMR (400 MHz, CDCl3): δ 1.80 (m, 8H), 1.71 (m, 4H), 1.50 (m, 4H), 1.25 (m, 20H), 0.86 (t, 3JHH = 6.9 Hz, 3H), 0.29 (q, 1JHB = 98 Hz, 3H); 13C NMR (101 MHz, CDCl3): δ 32.2 (s), 32.1 (d, 3JCP = 12.8 Hz), 32.0 (d, 1JCP = 32.6 Hz), 31.9 (d, 1JCP = 32.6 Hz), 29.55 (s), 29.5 (s), 29.45 (s), 29.4 (s), 27.35 (s), 27.32 (s), 27.30 (s), 27.28 (s), 27.22 (d, 3JCP = 15.7 Hz), 27.20 (d, 3JCP = 14.2 Hz), 26.42 (s), 26.40 (s), 24.1 (s), 23.0 (s), 19.8 (d, 1JCP = 32.2 Hz), 14.5 (s); 11B NMR (128 MHz, CDCl3): δ –40.1. HRMS calcd for C20H39P ([M–BH3]+•) 310.2789, found 310.2775.
2.3.6 Experimental procedure for 9
Method B: methyl-phenylphosphine borane 8 (115 mg, 0.83 mmol), oct-1-ene (157 μl, 1 mmol), toluene (1 ml), t = 65 h, T = 60 °C. The crude product was then purified by silica-gel column chromatography with toluene as eluent (Rf = 0.7). Phosphine borane 9 was obtained as a colourless oil in 62% yield. 31P NMR (162 MHz, CDCl3): δ 9.0; 1H NMR (400 MHz, CDCl3): δ 7.74 (m, 2H), 7.50 (m, 3H), 1.85 (m, 2H), 1.56 (d, 2JHP = 10.2Hz, 3H), 1.55–1.22 (m, 12H), 0.88 (t, 3JHH = 6.8 Hz, 3H), 0.79 (q, 1JHB = 98 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 131.7 (d, 2JCP = 9.6 Hz), 131.5 (d, 4JCP = 2.3 Hz), 130.3 (d, 1JCP = 53.3 Hz), 129.1 (d, 3JCP = 9.7 Hz), 32.1 (s), 31.4 (d, 3JCP = 13.2 Hz), 29.5 (s), 29.4 (s), 27.8 (d, 1JCP = 36.5 Hz), 23.4 (s), 23.0 (s), 14.4 (s), 11.1 (d, 1JCP = 39.0 Hz); 11B NMR (128 MHz, CDCl3): δ –36.4; HRMS calcd for C15H25P ([M–BH3]+•) 236.1694, found 236.1713.
2.3.7 Experimental procedure for 10
Method B: diphenylphosphine borane 1 (140 mg, 0.7 mmol), cyclohexene (85 μl, 0.84 mmol), toluene (1.2 ml), t = 72 h, T =RT. The crude product was then purified by silica-gel column chromatography with toluene as eluent (Rf = 0.7). Phosphine borane 10 was obtained as a white powder in 79% yield (mp = 92 °C). 31P NMR (101 MHz, CDCl3): δ 22.3; 1H NMR (250 MHz, CDCl3): δ 7.73 (m, 4H), 7.44 (m, 6H), 2.25 (dtt, 2JHP = 13.9 Hz, 3JHH = 11.9 Hz, 3JHH = 3.0 Hz, 1H), 1.78 (m, 2H), 1.66 (m, 3H), 1.45 (m, 2H), 1.25 (m, 3H), 0.95 (q, 1JHB = 98 Hz, 3H); 13C NMR (63 MHz, CDCl3): δ 132.8 (d, 2JCP = 8.2 Hz), 131.1 (d, 4JCP = 2.5 Hz), 128.8 (d, 3JCP = 9.4 Hz), 129.2 (d, 1JCP = 53.6 Hz), 33.9 (d, 1JCP = 36.0 Hz), 26.8 (d, 3JCP = 12.3 Hz), 26.6 (s), 25.9 (d, 4JCP = 1.3 Hz); 11B NMR (128 MHz, CDCl3): δ –36.6; HRMS calcd for C18H20P ([M–BH3]+•) 268.1381, found 268.1382.
2.3.8 Experimental procedure for 12
Method A: diphenylphosphine borane 1 (100 mg, 0.5 mmol), (–)-β-pinene (88 μl, 0.55 mmol), toluene (1 ml), t = 30 min, T = 70 °C. The crude product was then purified by silica-gel column chromatography with dichloromethane as eluent (Rf = 0.8). Phosphine borane 12 was obtained as a white solid in 95% yield (mp = 74 °C). [α]D = –40.3 (toluene); 31P NMR (121 MHz, CDCl3): δ 15.5; 1H NMR (300 MHz, CDCl3): δ 7.60 (m, 4H), 7.30 (m, 6H), 5.15 (m, 1H), 2.85 (dm, 2JHP = 12.9 Hz, 2H), 1.85 (m, 2H), 1.80 (m, 1H), 1.55 (m, 2H), 1.30 (m, 1H), 1.05 (m, 2H), 1.00 (q, 1JHB = 97 Hz, 3H), 0.75 (d, 3JHH = 6.7 Hz, 3H), 0.70 (d, 3JHH = 6.7 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 132.7 (d, 2JCP = 8.6 Hz), 132.3 (d, 2JCP = 8.6 Hz), 131.2 (d, 4JCP = 1.4 Hz), 131.0 (d, 4JCP = 1.4 Hz), 130.1 (d, 1JCP = 53.7 Hz), 129.7 (d, 1JCP = 53.7 Hz), 128.7 (d, 2JCP = 5.6 Hz), 128.6 (d, 3JCP = 9.7 Hz), 128.3 (d, 3JCP = 9.7 Hz), 127.7 (d, 3JCP = 9.6 Hz), 39.8 (s), 35.4 (d, 1JCP = 32.9 Hz), 32.3 (s), 31.4 (d, 3JCP = 1.7 Hz), 29.6 (s), 26.7 (s), 20.4 (s), 20.1 (s); 11B NMR (96 MHz, CDCl3): δ –38.8; IR (KBr) 2358 (νnBH), 2340 (νnBH); HRMS calcd for C22H30BP ([M]+•) 336.2172, found 336.2179.
2.3.9 Experimental procedure for 13
Method A: t-butylphenylphosphine borane (100 μl, 0.5 mmol), (–)-β-pinene (96 μl, 0.6 mmol), t = 1 h, T = 80 °C. The crude product was then purified by silica-gel column chromatography with toluene as eluent (Rf = 0.80). Phosphine borane 13 was obtained as a mixture of two diastereomers in a 1:1 ratio (white solid, 90% yield, mp = 78 °C). 31P NMR (121 MHz, CDCl3): δ 30.0; 1H NMR (300 MHz, CDCl3): δ 7.70 (m, 2H), 7.35 (m, 3H), 5.35 (m, 1H), 2.90 (m, 1H), 2.50 (m, 1H), 1.90 (m, 2H), 1.80 (m, 1H), 1.55 (m, 2H), 1.25 (m, 1H), 1.05 (d, 3JHP = 13.4 Hz, 9H), 1.00 (m, 1H), 0.75 (d, 3JHH = 6.7 Hz, 3H), 0.70 (d, 3JHH = 6.7 Hz, 3H), 0.70 (q, 1JHB = 101 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 133.8 (d, 2JCP = 8.5 Hz), 131.0 (d, 4JCP = 2.7 Hz), 130.9 (d, 2JCP = 5.0 Hz), 130.6 (d, 2JCP = 5.0 Hz), 128.0 (d, 3JCP = 9.3 Hz), 126.9 (s), 126.8 (s), 126.8 (d, 1JCP = 47.1 Hz), 39.3 (s), 32.2 (s), 32.1 (s), 31.1 (d, 3JCP = 1.8 Hz), 31.0 (d, 3JCP = 1.8 Hz), 29.7 (d, 3JCP = 31.0 Hz), 29.3 (s), 28.2 (d, 1JCP = 30.6 Hz), 28.0 (d, 1JCP = 30.3 Hz), 26.3 (s), 26.2 (s), 25.6 (d, 2JCP = 2.3 Hz), 19.9 (s), 19.7 (s); 11B NMR (128 MHz, CDCl3): δ –38.3; IR (KBr) 2360 (νBH), 2340 (νBH); HRMS calcd for C20H34BP ([M]+•) 316.2491, found 316.2519.
2.3.10 Experimental procedure for 15
In a flamed nitrogen-flushed two-necked round bottomed flask was added sodium hydride, 60% in mineral oil (105 mg, 2.62 mmol). The hydride was washed with pentane (2 × 2 ml) and dried. THF was introduced, the solution cooled to –78 °C and 1 (500 mg, 2.5 mmol) added. The reaction was vigorously shaken and the temperature allowed to reach RT. After 2 h, the mixture was cooled to –10 °C and deuterated water (4 ml, 222 mmol) was injected for 30 min. After 1 h 30, the solution was extracted with toluene (2 × 7 ml) and the organic layers dried over deoxygenated magnesium sulphate. Evaporation of toluene afforded 15 as a white solid, in 90% yield. The purity reached 90% and the compound was used such without further purification. 31P NMR (121 MHz, C6D6): δ 0.0; 1H NMR (400 MHz, C6D6): δ 7.65 (m, 4H), 7.05 (m, 6H), 0.95 (q, 1JHB = 97 Hz, 3H); 13C NMR (101 MHz, C6D6): δ 133.3 (d, 2JCP = 9.4 Hz), 131.6 (d, 4JCP = 2.3 Hz), 129.2 (d, 3JCP = 10.2 Hz), 122.3 (d, 1JCP = 56 Hz); 11B NMR (96 MHz, CDCl3): δ –36.3.
2.3.11 Experimental procedure for 18
Method A: deuterated diphenylphosphine borane (101 mg, 0.5 mmol), (–)-β-pinene (88 μl, 0.55 mmol), toluene (1 ml), t = 30 min, T = 70 °C. The crude product was then purified by silica-gel column chromatography with dichloromethane as eluent (Rf = 0.8). Phosphine borane 18 was obtained as a white solid in 95% yield. 31P NMR (121 MHz, CDCl3): δ d 15.5; 1H NMR (300 MHz, CDCl3): δ 7.60 (m, 4H), 7.30 (m, 6H), 5.15 (m, 1H), 2.85 (dm, 2JHP = 12.9 Hz, 2H), 1.85 (m, 2H), 1.80 (m, 1H), 1.55 (m, 2H), 1.05 (m, 2H), 1.00 (q, 1JHB = 97 Hz, 3H), 0.75 (s, 3H), 0.70 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 132.7 (d, 2JCP = 8.6 Hz), 132.3 (d, 2JCP = 8.6 Hz), 131.2 (d, 4JCP = 1.4 Hz), 131.0 (d, 4JCP = 1.4 Hz), 130.1 (d, 1JCP = 53.7 Hz), 129.7 (d, 1JCP = 53.7 Hz), 128.7 (d, 2JCP = 5.6 Hz), 128.6 (d, 3JCP = 9.7 Hz), 128.3 (d, 3JCP = 9.7 Hz), 127.7 (d, 3JCP = 9.6 Hz), 39.6 (s), 35.4 (d, 1JCP = 32.9 Hz), 31.8 (s), 31.4 (d, 3JCP = 1.7 Hz), 29.6 (d, 4JCP = 2.3 Hz), 26.7 (s), 20.3 (s), 20.0 (s); 11B NMR (96 MHz, CDCl3): δ –38.8.
3 Results and discussion
3.1 Microwave-assisted hydrophosphination (method A)
To get an impression of the reactivity of secondary phosphine boranes with unactivated alkenes, we first conducted experiments to assess the feasibility of the addition of diphenylphosphine borane 1 onto oct-1-ene. Similar to our recent work on hydrophosphination of alkynes [25], we performed the reaction under microwave irradiation, a technology that has gained considerable attention over past decades [29–30]. The alkene (5 equiv) and the phosphine borane 1 (1 equiv) were mixed together and heated at 50 °C. No catalyst was required and the reaction was conducted without solvent. After completion (30 min), analysis of the crude product by 1H NMR indicated that the hydrophosphination reaction proceeded in an anti-Markovnikov fashion, the phosphorus atom attacking exclusively the terminal carbon atom of the unsaturated compound as already observed with alkynes [25] (Scheme 1).
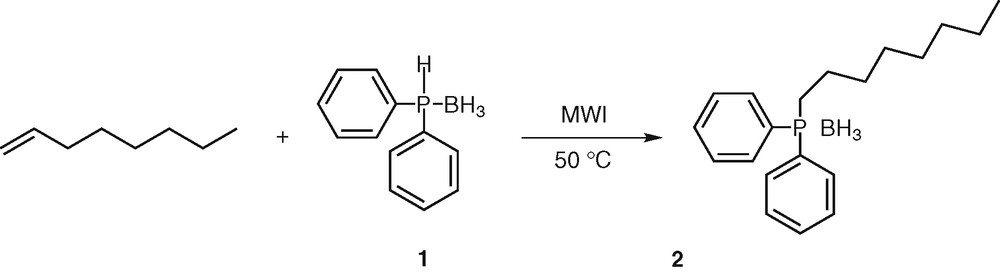
Hydrophosphination of oct-1-ene.
Decomplexation and oxidation of the phosphine derivatives were not observed, even though inert gas was not used. Octyldiphenylphosphine borane 2 is an air-stable compound, which was isolated after purification by silica gel chromatography in 68% yield. This preliminary result is to compare with the one obtained by Imamoto [26] using the same substrate and reactant under radical activation (35% yield). It indicates that the use of such a catalyst does not favour the reaction, the microwave-assisted hydrophosphination being a more reliable process (68% yield).
Our aim being to develop a route that would allow maximum flexibility for the variation of the phosphine moiety and for the alkene scaffold, hydrophosphination reactions of unactivated alkenes with various sterical hindrance was next investigated (Table 1, Scheme 2). Under the same conditions, the reaction was effective for hex-1-ene, and even for the sterically crowded 2,3,3-trimethylbutene, though the latter reacted sluggishly. Purification by column chromatography on silica gel afforded the desired phosphines 4 and 5 in 70 and 64% yields respectively (Scheme 2). Only the anti-Markovnikov adduct was obtained each time. Unactivated alkene carrying a silyl function that can allow further transformations [31] proved also to be a valuable substrate for the hydrophosphination reaction. The addition proceeded to completion at 50 °C in 25 min (Scheme 2). Phosphine 6 was obtained in 60% yield after purification by silica-gel chromatography.
Microwave activated hydrophosphination of alkenes
| Phosphine borane (1 equiv.) | Alkene (5 equiv) | Temperature (°C) | Time (min) | Product | Isolated yield (%) |
| 1 | oct-1-ene | 50 | 30 | 2 | 68 |
| 1 | hex-1-ene | 50 | 30 | 4 | 70 |
| 1 | 2,3,3-trimethylbutene | 50 | 30 | 5 | 64 |
| 1 | allyltrimethylsilane | 50 | 25 | 6 | 60 |
| 3 | oct-1-ene | 80 | 50 | 7 | 58 |
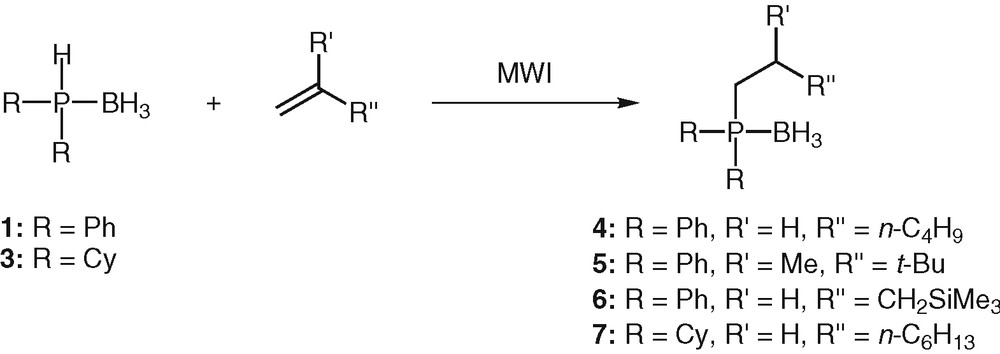
Microwave-activated hydrophosphination of alkenes.
In general, the tertiary phosphines were produced in reasonable to high chemical yields (58 to 70% yields). However, side products were detected in varying degree of abundance (2 to 15%), depending on the temperature and on the length of the reaction. They mainly resulted from the decomplexation of the phosphine boranes, followed by their oxidation. The competitive hydroboration process was never observed.
To validate the generality of the method, we next turned our attention to the less reactive dialkylphosphine boranes. Dicyclohexylphosphine borane 3, a highly electron-rich and bulky secondary phosphine borane was engaged in hydrophosphination reaction with oct-1-ene (Scheme 2). Not unexpectedly, the reaction required longer reaction time (50 min) and higher temperature (80 °C) compared to the one involving diphenylphosphine borane 1. This lower reactivity is attributable to the steric effect of the substituents (cyclohexyl group) but also to the strongest P–H bond (sigma donor effect of the cyclohexyl groups). Nevertheless, the yield of the reaction was satisfactory, phosphine 7 being obtained in 58% yield after purification by silica-gel column chromatography. All the results of the microwave induced alkenes hydrophosphination are summarized in Table 1. Because the microwave devices are not commonly available, we sought to perform the reactions using conventional heating in oil baths.
3.2 Hydrophosphination under conventional heating (method B)
The reaction between oct-1-ene (1.2 equiv) and diphenylphosphine borane 1 (1 equiv) was chosen as a model reaction. Results from preliminary experiments using several solvents showed satisfactory conversion with toluene at 60 °C. The addition was monitored by TLC until 1 was consumed. Under these conditions, the reaction was still progressing after 12 h and was essentially complete after 20 h. The hydrophosphination product obtained was readily identified as the anti-Markovnikov adduct 2. The isolated yield is moderate (52%). It is necessary to point out that the reaction can even be performed at room temperature, even though it takes a longer time. After 48 h, the 31P NMR spectrum showed a clean formation of the desired product. After silica gel chromatography, the phosphine borane 2 was isolated in pure form in 69% yield. Upscaling the reaction to 1 gram of 1 did not change the efficiency or the yield of the reaction. These exceptionally mild hydrophosphination conditions (absence of catalyst, room temperature) may be explained by the strong P–H activation induced by the borane group. Other alkenes such as hex-1-ene and allyltrimethylsilane also reacted successfully under the same conditions at room temperature with 1. The reactions were completed respectively after 72 and 65 h at RT (Scheme 3, Table 2). The desired products 4 and 6 were obtained in 65% and 62% yields respectively.
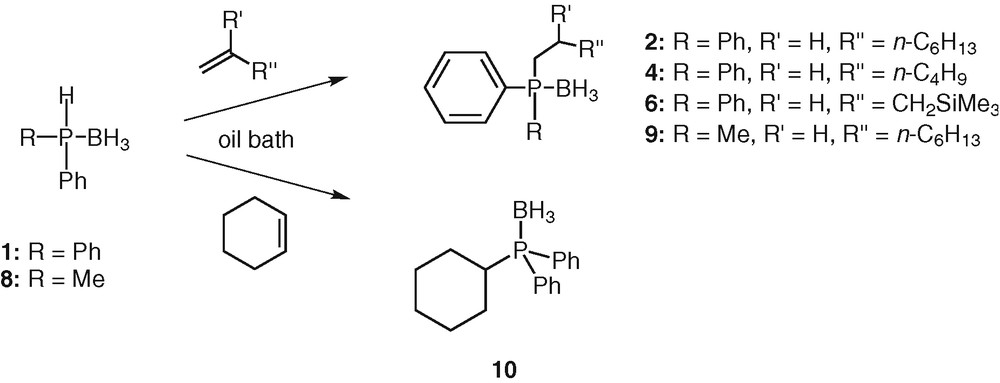
Thermal hydrophosphination.
Thermal activated hydrophosphination of alkenes
| Phosphine borane (1 equiv) | Alkene (1.2 equiv) | Temperature (°C) | Time (h) | Product | Isolated yield (%) |
| 1 | oct-1-ene | 60 | 20 | 2 | 52 |
| 1 | oct-1-ene | RT | 48 | 2 | 69 |
| 1 | hex-1-ene | RT | 72 | 4 | 65 |
| 1 | Allyltrimethylsilane | RT | 65 | 6 | 62 |
| 1 | Cyclohexene | RT | 72 | 10 | 79 |
| 8 | oct-1-ene | 60 | 65 | 9 | 62 |
Even internal alkene such as cyclohexene could be hydrophosphinated using these conditions. The reaction between diphenylphosphine borane 1 and cyclohexene was completed after 72 h in toluene at RT. Purification by silica-gel chromatography afforded 10 in 79% yield (Scheme 3). This result compared to the 7% yield obtained by Imamoto [26] using the same substrate and reactant under radical conditions confirmed that radical initiators are less beneficial than thermal activation to add a phosphine borane across an unactivated alkene.
The reaction was also applicable to alkylarylphosphine boranes but required a gentle heating. At room temperature, methylphenylphosphine borane 8 reacted with oct-1-ene in toluene even though it appeared that the reaction was sluggish in comparison with the one involving diphenylphosphine borane 1. After one week, only traces of the desired product 9 were observed. Significantly better results were obtained upon heating the solution at 60 °C in an oil bath. The addition reaction was completed within 65 h (Scheme 3). The reaction was again completely regioselective, giving only the anti-Markovnikov adduct 9. The yield of isolated product after silica gel chromatography was quite reasonable (62%). The lowest reactivity of methylphenylphosphine borane 8 compared to diphenylphosphine borane 1 is attributable to the strongest P-H bond (sigma donor effect of the methyl group).
3.3 Hydrophosphination of chiral alkenes
Using a chiral alkene derived from the chiral pool (e.g., a terpene) should open the way to the preparation of chiral phosphines. We thus reacted diphenylphosphine borane 1 with (–)-β-pinene. Heating the mixture neat at 70 °C for 30 min under microwave irradiation followed by purification by silica gel chromatography afforded enantiopure phosphine borane 12 in 95% yield. Phosphine 14 was never observed (Scheme 4). With a bulky and electron rich secondary phosphine borane (e.g., tert-butylphenylphosphine borane 11), the same result was observed, leading to 13 in 90% yield. Due to the chiral phosphorus atom, phosphine 13 was obtained as a mixture of two diastereomers in a 1:1 ratio, which were not separated (Scheme 4).
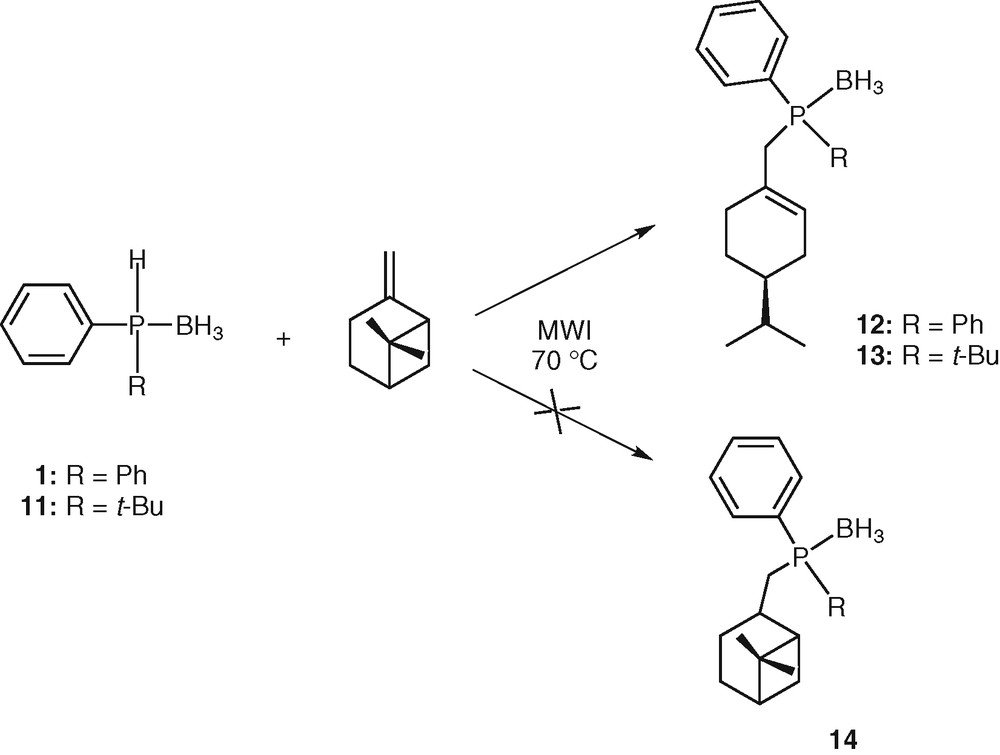
Hydrophosphination of pinene.
The formation of 12 could be explained by a radical process involving the following reaction sequences: (i) homolytic cleavage of the P–H bond giving the phosphinyl radical [H3B•PPh2]• 16, (ii) addition of the phosphinyl radical 16 to alkene leading to an alkyl radical, (iii) rearrangement of the alkyl radical by bridge opening [32], (iv) abstraction of an hydrogen atom from another phosphine borane 1 by the alkyl radical, leading to the final product 12 (Scheme 5). This ring opening would be thermodynamically favoured, since leading to a less strained molecule.
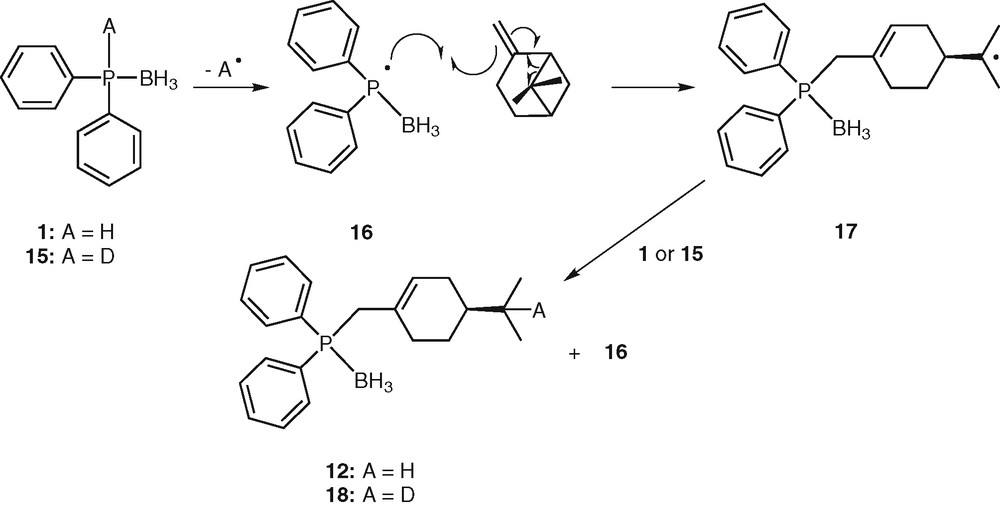
Proposed mechanism for hydrophosphination of β-pinene.
In order to obtain some mechanistic information, the reaction was performed with 1-deutero-diphenylphosphine borane 15. The latter was formed by deprotonation of diphenylphosphine borane 1 with NaH, followed by hydrolysis with D2O. The NMR analysis of the hydrophosphination product unambiguously showed the addition of deuterium atom on radical 16, leading exclusively to 18. This observation strengthens our belief that a radical mechanism is at least partially involved in the hydrophosphination process. Nevertheless, further investigation should be carried out to ensure this proposal.
4 Conclusion
In summary, the present work demonstrates an efficient means for rapidly and easily generating P-alkyl substituted tertiary phosphines from commercially available alkenes and secondary phosphine boranes. The borane group acts as a protecting group towards oxidation and furthermore activates the P–H bond, making secondary phosphine boranes suitable phosphination agents. Thus, the hydrophosphination proceeded smoothly under conventional heating or under microwave irradiation, though a marked acceleration rate was observed in the microwave reactions. Clearly, this hydrophosphination process is atom economic and environmentally friendly. The starting alkenes are both inexpensive and readily available. The methodology outlined provides a rapid access to P-alkylphosphines and paves the way to the preparation of new phosphine families.
Acknowledgments
This work has been performed within the ‘PUNCHOrga’ interregional network (‘Pôle universitaire de chimie organique’). We gratefully acknowledge financial support from the French ‘Ministère de la Recherche et des Nouvelles Technologies’, CNRS (‘Centre national de la recherche scientifique’), the ‘Région Basse-Normandie’ and the European Union (FEDER funding) for financial support. D.M. acknowledges the French Ministry of Education and Research for a PhD fellowship. The authors also thank Prof. D. Villemin (ENSICAEN) for the loan of the MWI device.