

1 Introduction
Carotenoids are polyene hydrocarbons biosynthesized from eight isoprene units and, correspondingly, have a 40-C skeleton. They provide the intensive yellow, orange or red colour of a great number of plant-based foodstuffs. Synthesized principally by plants, they are generally present as a complex mixture.
Because they are very soluble in apolar solvents, but not in water, carotenoids are called ‘lipochromes’. Their colour is the result of the presence of a conjugated double bond system in the molecules. Carotenoids have been the subject of many scientific studies in the food industry, mostly because many of them are vitamin A precursors [1–4]. They are highly sensitive to oxygen and light. However, their degradation is accelerated by intermediate radicals occurring in food due to lipid peroxidation. The often-observed changes in colour intensity are related to the oxidative degradation of carotenoids. Aroma compounds are formed during the oxidative degradation of carotenoids. It is also known that carotenoids can lead to the formation of norisoprenoids, compounds that possess distinct aromatic characteristics through oxidative degradation catalysed by light [5] or coupled oxidation [6].
The presence of β-carotene and lutein in wood was first described by Masson et al. [7]. These compounds are found in the wood of sessile oak, pedunculate oak, American white oak, chestnut and beech. Amounts are generally small and vary considerably from one tree to another. Pinkish woods are considered to be the best for cooperage purposes. The molecules responsible for this pinkish hue have been identified and belong to the carotenoids family, principally β-carotene and lutein. This feature can be put to use in barrel making. Indeed, as the wood is exposed to light and oxidation during natural seasoning, and then to heat when the barrels are made, new aromatic compounds, some of them offering fruity and floral notes, can be produced during the various stages of barrel making. This kind of molecule participates in the aromatic and fruity character of oak wood and consequently enhances the gustatory qualities of wine during ageing in barrels.
Many studies have looked at how oak extracts contribute to the flavour of barrel-aged alcoholic beverages. Over 200 oak wood volatile compounds have been identified, and the aromatic and flavouring properties of some have been characterized [8–10].
In this paper, we first quantify carotenoids in different pieces of wood, and redefine carotenoid degradation factors; we then tentatively identify their degradation products using different methods. And finally, we describe the extraction method, the characterization conditions and the identification of 13-, 11-carbon norisoprenoids in Quercus petraea wood. Previously, Sefton et al. [11] presented an extraction method using freon as solvent. To isolate norisoprenoids, we perfected a method studying different factors such as maceration time in a hydroalcoholic solution, temperature, extraction solvent, GC/MS conditions. We thus identified 14 norisoprenoids in a sample of Q. petraea oak wood; most of them are not of the same nature.
2 Materials and methods
2.1 Quantification of carotenoids in oak wood
2.1.1 Sampling
Samples of French oak (Quercus petraea L.) were taken from several trees and from the same positions in the trunk, according to the difference of colour. A 15-cm-thick slice of wood was cut 2 m from the base of the tree trunk. Six samples were studied: freshly sawed brown and light brown wood, dry yellow wood, pink wood, pink + wood, pink ++ wood. Shavings were ground to a particle size of less than 0.5 mm and kept at –20 °C before use.
2.1.2 Extraction of carotenoids from wood
The techniques used for carotenoid extraction and assay methods were based on those developed by Razungles [12] for grape berries and applied by Masson et al. [7] Fresh wood powder (10 g) was mixed with magnesium carbonate (3 g) to neutralize the free acids in the wood that might cause isomerisation reactions in the carotenoids and was mixed with antioxidants: butylhydroxyanisol (BHA) and butylhydroxytoluene (BHT) to prevent carotenoid degradation. A known quantity of β-apo-8'-carotenal (internal standard) was added to this mixture. Carotenoids were extracted from the wood by 6 × 30 ml of acetone. The acetone extracts were separated by filtration and evaporated to dryness in a rotavapor. The residue was saponified: it was added to an ethanol:potassium hydroxide mixture (50 ml of ethanol + 5 ml of 60% KOH in water). The mixture was left for 12 h at 4 °C (to eliminate lipids and to precipitate polyphenols in the alcoholic phase). The saponified mixture was then placed in a separating funnel with 50 ml of ethyl ether. The ether phase was washed with water, dried over anhydrous sodium sulphate, and evaporated in a rotary evaporator to dryness. The residue was resuspended in 2 ml of acetone. The final extract obtained (containing wood carotenoids) was filtered before HPLC injection. It should be noted that all these operations were performed in the shade with no direct light, and at room temperature.
2.1.3 HPLC assay of carotenoids
The carotenoids were analysed with a set of Waters™ HPLC apparatus: a 486 MS multiwave length detector, 600 S controller, and a manual injector. A binary gradient was used. Solvent A was acetone/water (70:30). Solvent B was acetone. Flow-rate was 1 ml/min with a gradient of 0–100% B in 20 min, 100% B in 10 min, 100–0% B in 7 min, and 10 min of re-equilibration under the initial conditions. A C18 reverse phase column (250 mm × 4.6 mm, 10 μm) was used. The carotenoids were detected at 450 nm.
2.2 Pyrolysis/GC/MS of carotenoids
A ‘Pyrojector II’ was used in conjunction with a Varian™ 3400 CX model gas chromatograph equipped with an ion trap, Varian™ Saturn 4D detector. The samples were placed in the pyrolyser and duplicate pyrolysis experiments were carried out at 400 °C. Separation of the pyrolysis products was achieved in a fused-silica capillary column: J&W DB-5MS™ (30 m × 0.25 mm i.d. × 0.25 μm phase thickness). Helium was used as carrier gas. The inlet mode was splitless. The gas chromatograph was programmed from 60 (1 min) to 300 °C (20 min) at the rate of 4 °C min–1. The mass spectrometer was set at 70 eV. Mass spectra were scanned in the range m/z = 58–650. Spectra were acquired by a ChemStation software package. Identification was achieved by mass fragmentometry, a library search (NIST) and comparison with literature data.
2.3 Oxidation of β-carotene by copper and radical oxidation
2.3.1 Oxidation by copper
A few milligrams of carotenoids were introduced into 10 ml of a hydroalcoholic solution (ethanol 120 ml, tartaric acid 5 g, pH adjusted to 3.4 by 1 N NaOH, the final volume is adjusted to 1000 ml with distilled water) containing 2.5 mg l–1 copper or iron. After five days at room temperature, organic compounds were extracted by liquid–liquid extraction.
2.3.2 Radical oxidation
In a tube, we introduced 4.4 mg carotenoids, 2.453 ml trihydroxymethylaminomethane (TRIS, pH = 7), 25 μl xanthin and 500 μl hypoxanthin. After 15 min at room temperature, the reaction was stopped by adding acetic acid. Organic compounds were extracted by liquid–liquid extraction.
2.3.3 Extraction of degradation products
The filtrated solution was placed in a separating funnel and 1 g of sodium hydrogencarbonate was added. The solution was extracted three times with pentane; the organic phases were combined and dried over anhydrous sodium sulphate. The final phase was concentrated in a rotary evaporator to a final volume of 500 μl before GC/MS.
MS spectra were recorded by coupling a Varian™ 3400 CX model gas chromatograph equipped with a J&W DB-5MS™ (95% dimethyl, 5% diphenyl polysiloxane; 30 m × 0.25 mm i.d.; 0.25 μm bonded phase; J&W Scientific™) to an Varian™ Saturn 4D ion trap, mass spectrometer. The column temperature was programmed at 60 °C (1 min) from 310 °C at a rate of 4 °C min–1 (20 min). The temperature injector was 250 °C; 2 μl were injected in splitless mode. Detection was accomplished operating in electron mode with an ionisation energy of 70 eV with transfer-line temperature at 250 °C. Mass spectra were scanned in the m/z = 58–650 range. Identification was achieved by mass fragmentometry, a library search (NIST) and comparison with literature data.
2.4 Isolation and identification of volatile compounds and norisoprenoids: optimising the method
2.4.1 Preparation of wood samples
Oak wood samples were taken from naturally seasoned wood staves, by planning the wood and reducing it to sawdust (1–2 mm).
2.4.2 Extraction of wood samples
To reproduce extraction conditions similar to those in wines, the wood samples were placed in a hydroalcoholic model solution. A 100-g sample of sawdust was mixed with 1250 ml of a diluted alcohol solution (120 ml ethanol, 5 g tartaric acid, pH adjusted to 3.4 by 1 N NaOH, the final volume is adjusted to 1000 ml with distilled water), kept x days at 20 °C and mixed daily. The juice obtained was filtered.
First, the pH of the model solution was adjusted to 7 by dissolving 5 g of sodium hydrogencarbonate in 200 ml of the solution. The model solution was then extracted four times (500 ml, 360 ml, 240 ml, 120 ml) with different solvents: pentane, chloroform, dichloromethane, ethyl acetate, diethyl ether.
The resulting organic phase was dried, in each case, with anhydrous sodium sulphate, then concentrated on a rotary evaporator to a final volume of 1 ml. Each fraction was analysed by GC/MS as in conditions 1 and 2.
2.4.3 Analysis of the fractions by GC/MS (optimisation of analysis conditions)
MS spectra were recorded by coupling a Hewlett-Packard (HP)™ 5890 series II gas chromatography, equipped with a J&W DB-5MS™ or Carbowax™ column, to a VG-Autospec EQ™ mass spectrometer.
Conditions 1
GC analyses were performed on a HP Innowax™ column (100% polyethylenglycol; 30 m × 0.25 mm i.d.; 0.25 μm bonded phase). The column temperature was programmed at 35 °C (50 s) from 45 °C at a rate of 50 °C min, from 240 °C at a rate of 3 °C min -1 (30 min) with helium as carrier gas. The temperature injector was 220 °C; 2 μl were injected in splitless mode. Detection was accomplished operating in electron mode with an ionisation energy of 70 eV and the transfer line was heated at 220 °C. Mass spectra were scanned in the range m/z = 58–850.
Conditions 2
GC analyses were performed on a DB-5MS™ column (95% dimethyl, 5% diphenyl polysiloxane; 30 m × 0.25 mm i.d.; 0.25 μm bonded phase; J&W Scientific™). The column temperature was programmed at 60 °C from 310 °C at a rate of 4 °C min (30 min). The temperature injector was 280 °C; 2 μl were injected in splitless mode. Detection was accomplished operating in electron mode with an ionisation energy of 70 eV and transfer-line temperature was 280 °C. Mass spectra were scanned in the m/z = 58–850 range.
3 Results and discussion
3.1 Quantification of carotenoids in wood
Masson et al. [7] demonstrated that two carotenoids, β-carotene and lutein (Fig. 1), are found in oak wood. They could come from leaves, in which they are synthesized in large quantities, or the second hypothesis is that of the in situ formation of carotenoids in the living cells of the sapwood, as a protection against singlet oxygen, as reviewed by Larson [13].
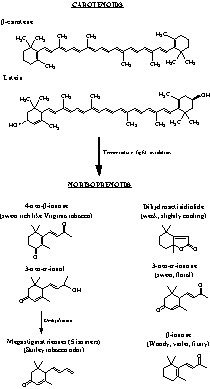
Structure of β-carotene, lutein and characteristics of some of their degradation products.
Fig. 2 represents HPLC chromatogram of carotenoids extracted from oak wood. Carotenoid identification was performed by comparing their retention times with those of reference substances. Table 1 sums up the results obtained from different coloured woods. The colour of oak wood has a readily detectable effect on the content of this carotenoid. Pink woods are richer in carotenoids than freshly sawed brown or yellow woods. The darker the pink (pink, pink+, pink++), the greater the carotenoid content. This type of wood is consequently considered to be the best for making a new kind of selected barrels.
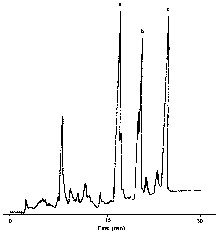
HPLC chromatogram of carotenoids extracted from oak wood. Peak a: lutein; Peak b: β-carotenal; Peak c: β-carotene.
Lutein (Lut) and β−carotene (β−car) contents (μg/g dry weight of wood) and total contents (Lutt+ β−car) in mg/g dry weight of wood
| Colour of wood | Lut | β−car | Lut+β-car |
| Freshly sawed brown | 0.96 | 0.19 | 1.15 |
| Light brown | 0.18 | 0.38 | 0.56 |
| Dry yellow | 0.09 | 0.22 | 0.31 |
| Pink | 0.88 | 0.28 | 1.16 |
| Pink+ | 0.95 | 0.54 | 1.49 |
| Pink++ | 1.2 | 1.1 | 2.3 |
3.2 Factors generating carotenoid degradation
Only β-carotene was used to study the influence of different parameters on carotenoid degradation. Light, temperature and oxidation are the three factors mentioned in the literature by different authors [5,6,14,15]. The experimental part was very simple: five solutions at 2 mg l–1 of β-carotene were prepared. With each one, the effect of light, temperature and oxidation on the degradation of β-carotene was studied. The first tube was saturated with nitrogen and placed in normal daylight. To study the influence of temperature, three tubes saturated in nitrogen and sheltered from light were placed at room temperature, at 40 and 70 °C. The last tube, placed in the dark at 0 °C, was oxidized by copper [10 ml solution of β-carotene at 2 mg l–1 (hydroalcoholic solution containing 2.5 mg l–1 copper or iron)]. In each case, the kinetics of the disappearance of β-carotene was assessed by HPLC (see experimental part for the ‘HPLC assay of carotenoids’ conditions).
Fig. 3 sum up the results obtained in each case. Under the influence of light, we noted that β-carotene disappeared very quickly; after 30 min, its concentration is divided by 2. At room temperature (in the dark), β-carotene deteriorates very slowly; but, more the temperature increases and more this degradation speeds up. For high temperatures (T ≥ 70 °C), the degradation is instantaneous. In the same way, oxidation (at 0 °C and in the dark) is another parameter that induces a rapid degradation of β-carotene.
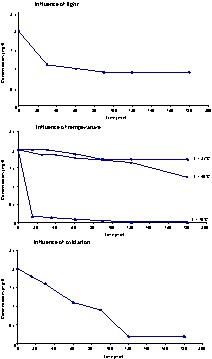
Influence of light, temperature and oxidation on the degradation of β-carotene.
3.3 Degradation products of β-carotene and lutein by pyrolysis and oxidation
Pyrolysis/Gas Chromatography/Mass Spectrometry (PY/GC/MS) is a useful tool to simulate the heating of barrels. PY/GC/MS is based on the degradation of molecules by heat. The increase in temperature leads to their thermal degradation and the products formed can then be identified [16]. Pyrolysis products are separated by gas chromatography and identified with mass spectrometry. Fig. 4 shows the total ion chromatogram of the thermal degradation products obtained after pyrolysis of β-carotene and lutein respectively. Many peaks were identified. In the case of PY/GC/MS of β-carotene (Fig. 4A), essentially two classes of compounds are recognizable, i.e. norisoprenoids and sesquiterpenes. Four megastigmatrienones and β-ionone were identified. A lot of sesquiterpenes are present; their identification was not achieved because their fragmentation patterns are similar (in each case, only the relative intensity varied). For our study, it was only important to identify norisoprenoids. PY/GC/MS of lutein (Fig. 4B) is characterized by the presence of terpenes and norisoprenoids. Two megastigmatrienones, 6-hydroxy-α-ionone, 2-hydroxy-β-ionone were identified as norisoprenoids.

Pyrolysis/GC/chromatograms of (A) β-carotene and (B) lutein. Peaks a–d: megastigmatrienones; peak e: β-ionone; peak f: 6-hydroxy-α-ionone; peak g: 2-hydroxy-β-ionone. * sesquiterpenes.
Two kinds of oxidation reactions were used to simulate this phenomenon in barrels: oxidation by copper and radical oxidation. They were applied to β-carotene. In the first case, we identified β-cyclocitral, β-ionone, dihydroactinidiolide, 3-oxo-β-ionone, 6-hydroxy-α-ionone and trans-β-iono-5,6-epoxide as degradation products (Fig. 5A). In the second case, the same compounds were characterized, 3-oxo-β-ionol and a lot of sesquiterpenes (Fig. 5B).

GC chromatogram of degradation products of β-carotene (A) oxidation by copper and (B) oxidation by radical oxidation. Peak a: β-cyclocitral; peak b: β-ionone; peak c: trans-β-iono-5,6-epoxyde; peak d: dihydroactinidiolide; peak e: 3-oxo-β-ionone; peak f: 6-hydroxy-α-ionone; peak g: 3-oxo-β-ionol. * Sesquiterpenes.
It is clear that two classes of compounds are produced when carotenoid degradation occurs (by pyrolysis or oxidation): norisoprenoids and sesquiterpenes. But, in our study, the most important was the presence of norisoprenoids as degradation products.
To isolate norisoprenoids in oak wood, we developed a specific method with first a maceration in hydroalcoholic solution and then with the best extraction and GC/MS analysis conditions. A great number of parameters were studied (extraction solvents, maceration time, analysis conditions in GC/MS) and are described below. But it is important to note that these parameters are not dissociable and can vary at the same moment.
3.4 Extraction and analysis conditions of norisoprenoids in oak wood
To find the best extraction solvent, we set a standard maceration time: 45 days at room temperature. We deliberately chose a long period of time so as to eliminate the influence of this parameter. Several solvents were tested according to the protocol described in the experimental section and each fraction obtained was analysed by both GC/MS methods (DB-5MS™ and HP INNOWAX™ columns). The pentane, ethyl acetate, diethyl ether, chloroform were rejected due to their non-specificity in the norisoprenoid extraction in oak wood. Among the different solvents tried and of the two conditions proposed (conditions named 1 and 2) for the GC/MS analysis, we retained dichloromethane, which proved to be the most suitable solvent for extracting norisoprenoids from oak wood and the first GC analysis conditions. Indeed, we noted that a polar column was the most suitable to separate norisoprenoids (good resolution and sensitivity); conversely, an apolar column gave full chromatograms with a poor resolution.
3.4.1 Influence of maceration time
Sawdust was put into contact with a hydroalcoholic solution for 45, 25, and 7 days. After 45 days, the GC chromatogram presented a lot of volatile phenols and 14 norisoprenoids. The same results were observed after 25 and 7 days. It thus appears that it takes only a few days to extract norisoprenoids in oak wood and these are quickly solubilised in wines and spirits.
3.4.2 Influence of temperature
Some assays were performed placing a hydroalcoholic solution at 55 °C for 7 days. The extraction solvent used was dichloromethane and compounds were analysed using a HP Innowax™ column. The GC chromatogram presented volatile compounds and on one occasion norisoprenoids (the same as those found above). The three norisoprenoids that were not characterized were certainly destroyed by heating the model solution. Extraction at room temperature proved to be the best method.
3.5 Identification of norisoprenoids in oak wood
With the GC/MS analysis in conditions 1 (See ‘Experimental’ section) norisoprenoids in Q. petraea extracts (obtained previously-after maceration in hydroalcoholic solution, and then extraction by organic solvent) were separated and identified. This method yielded novel results; for the first time, they revealed the presence of 14 norisoprenoids (Fig. 6). The capillary GC profile is presented in Fig. 7. Identification was achieved by mass fragmentometry (Fig. 8), a library search (NIST) and comparison with literature data, principally with references from Sefton et al. [11]. The norisoprenoids identified in the model wine extracts of Q. petraea oak samples are listed in Table 2, together with their chromatographic data (relative retention times), mass spectrometric data (molecular ions and characteristic fragments). EI-MS (Fig. 8) data for Blumenol A and 3-oxo-α-ionol were in good agreement with those published by Suarez [17] and by Aasen et al. [18].

Norisoprenoids observed in oak wood. No absolute stoichiometry is implied.
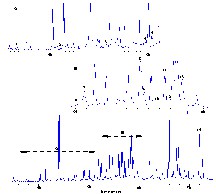
GC chromatogram of norisoprenoids identified in oak wood (corresponding numbers are listed in Table 2).
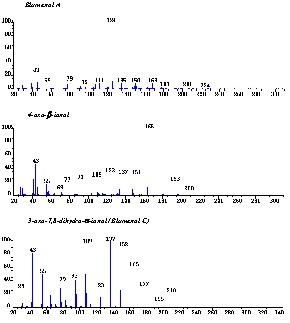
Mass spectrum of some identified norisoprenoids in oak wood.
Norisoprenoids identified in Quercus petraea and their principal fragmentations
| Peak | Products | EI fragments (relative intensity) | |||||||||||
| 1 | 7-oxo-dihydrotheaspirane | 210 (10) | 154 (85) | 139 (60) | 126 (60) | 112 (100) | |||||||
| 2 | megastigmatrienone | 190 (100) | 175 (87) | 148 (94) | 119 (55) | 105 (44) | 91(63) | 77(39) | 41(52) | ||||
| 3 | dihydroactinidiolide | 180 (25) | 165 (12) | 152 (30) | 137 (36) | ||||||||
| 4 | megastigma-4,7,9-trien-3-one | 190 (6) | 134 (100) | 119 (42) | 106 (23) | 91 (67) | 79 (12) | 65 (11) | 41 (13) | ||||
| 5 | 4-oxo-β-ionone | 206 (80) | 191 (12) | 163 (100) | 149 (25) | 135 (23) | 121 (40) | 107 (10) | 91 (13) | 43 (58) | |||
| 6 | 3-hydroxy-β-damascone | 208 (40) | 193 (35) | 175 (41) | 160 (6) | 149 (15) | 134 (6) | 121 (52) | 107 (12) | 95 (13) | 79 (10) | 69 (100) | 43 (43) |
| 7 | 3-oxo-7,8-dihydro-α-ionol | 208 (56) | 192 (8) | 177 (30) | 150 (60) | 135 (75) | 123 (30) | 108 (78) | 93 (72) | 84 (42) | 69 (77) | 55 (40) | 43 (100) |
| 8 | 3-oxo-α-ionol | 208 (2) | 193 (1) | 165 (2) | 152 (19) | 135 (6) | 123 (3) | 108 (100) | 95 (10) | 43 (27) | |||
| 9 | 4-oxo-β-ionol | 208 (1) | 193 (20) | 165 (100) | 151 (58) | 137 (40) | 122 (40) | 109 (38) | 91 (24) | 79 (20) | 55 (17) | 43 (40) | |
| 10 | dihydro-β-ionone | 194 (12) | 179 (38) | 161 (50) | 136 (40) | 121 (100) | 107 (18) | 93 (38) | 43 (62) | ||||
| 11 | dihydro-3-oxo- β-ionol | 210 (8) | 195 (10) | 177 (5) | 165 (20) | 152 (30) | 137 (40) | 121 (15) | 109 (32) | 95 (22) | 69 (23) | 55 (47) | 43 (100) |
| 12 | 2 hydroxy-β-ionone | 208 (10) | 193 (100) | 175 (53) | 149 (32) | 133 (9) | 121 (45) | 105 (25) | 91 (22) | 77 (11) | 55 (11) | 43 (96) | |
| 13 | blumenol C | 210 (56) | 192 (8) | 177 (30) | 150 (60) | 135 (75) | 123 (230) | 108 (78) | 93 (72) | 84(42) | 69 (77) | 55 (40) | 43 (100) |
| 14 | blumenol A | 224 (1) | 159 (5) | 124 (100) | 79 (10) | 43 (20) |
Most of the volatile norisoprenoids that were detected are C13 compounds. It is the first time that one of them has been identified in French oak wood. Several of these norisoprenoid oak constituents may well contribute to oak-derived flavours in wines and spirits. The presence of 3-oxo-α-ionol (8) is important: this compound, with just a faint smell, can lead, by dehydration and under mild acidic conditions [19,20] to five megastigmatrienone isomers known as the key flavour constituents in the aroma of Burley tobacco [21]; they essentially give a spicy, peppery note [22–24]. The mass spectra of four of the isomers are extremely similar, suggesting that these compounds differ only with respect to the configurations of the side-chain double bonds. In the extract, we identified two of the four isomers: compounds 2 and 4. These compounds have all been patented as flavour additives in the food, tobacco, and perfume industries.
The reduced form of 4-oxo-β-ionone (5) was detected in our sample as the degraded carotenoid: 4-oxo-β-ionol (9). 3-hydroxy-β-damascone (6) is a widespread industrial flavouring compound, so its presence in oak wood can be beneficial for wines and spirits ageing in oak barrels. Other norisoprenoids identified in Q. petraea have aromatic properties: dihydroactinidiolide (3), weak, slightly cooling; 4-oxo-β-ionone (5), rich and sweet, like Virginia tobacco.
4 Conclusion
Amounts of carotenoids in oak wood are generally small and vary considerably between samples, depending notably on the colour of the piece of wood. Pinkish oak woods are significantly richer in carotenoids than other coloured woods. These compounds are highly sensitive to oxygen, light and temperature, producing mostly norisoprenoids. Most of the volatile norisoprenoids that were detected in oak wood with the new method proposed, are C13 compounds. Several of these norisoprenoid oak constituents may well contribute to oak-derived flavours in wines and spirit.