

1 Introduction
Polyoxometalates (POMs) have been discovered by Berzelius [1] almost two centuries ago, but the first structural details were only revealed by Keggin [2] in the last century. POMs are composed of octahedrally coordinated transition metal atoms of groups V and VI in their highest oxidation states (e.g. V5+, W6+) [3]. The structural variety of POMs is due to sharing of these MO6 octahedra by edges and corners. Incorporation of one or more hetero groups (e.g. SiO4, AsO3) as additional building blocks allows for further structural versatility. Furthermore, substitution of one or more addenda atoms by redox-active transition metal ions (e.g., Fe3+, Ru3+) allows for fine tuning of the red-ox properties of POMs. Meanwhile this class of metal–oxygen clusters has generated substantial interest because of its unique structural variety and exciting properties (catalysis, magnetism, materials science and medicine) [4–12].
The synthesis of POMs is mostly rather simple and straightforward, once the proper reactions conditions have been identified. However, the mechanism of formation of POMs is not yet well understood and commonly described as self-assembly. Therefore, the design of novel POMs remains a challenge for synthetic chemists. The most rational synthesis procedure of POMs involves the use of lacunary precursors. Lacunary POMs are usually synthesized from complete precursor ions by loss of one or more MO6 octahedra. Reaction of a stable, lacunary POM with transition metal ions mostly leads to a product with the heteropolyanion framework unchanged. This approach usually results in monomeric or dimeric polyoxoanions with expected structures, e.g. [A-α-SiW9O34]10– + 3 Cu2+ → [Cu3(H2O)3(A-α-SiW9O37]10– and 2[B-α-PW9O34]9– + 4 Co2+ → [Co4(H2O)2(B-α-PW9O34)2]10– [13,14].
Sandwich-type POMs based on two [B-α-XW9O34]n– (X = PV, AsV, SiIV, GeIV) or [X2W15O56]12– (X = PV, AsV) fragments and four transition metal centers constitute a well-known class of compounds [15]. To date a large number of derivatives has been reported for both the Keggin- and Wells–Dawson-type structures. It has become apparent that this dimeric, sandwich-type structure (Weakley-type) allows for incorporation of essentially all first-row and even some second row transition metal ions [15].
Recently several examples of Weakley-type sandwich POMs with less than four transition metals have been reported [16]. Hill et al. described di- and tri-iron-substituted polyanions of the Wells–Dawson type and interestingly the vacancies were occupied by sodium ions (e.g., [Fe2(NaOH2)2(P2W15O56)2]16–, [Fe2(FeOH2)(NaOH2)(P2W15O56)2]14–). However, it has been possible to substitute these sodium ions by first-row-transition metal ions leading to mixed-metal sandwich-type polyanions [16]. The first example of a tri-substituted, sandwich-type polyoxoanion based on Keggin fragment is also known ([Ni3Na(H2O)2(B-α-PW9O34)2]11–) [17].
Lone pair containing polyoxotungstates (e.g. [Cs2Na(H2O)10Pd3(α-SbIIIW9O33)2]9–, [Cu4K2(H2O)8(α-AsIIIW9O33)2]8–), [AsIII6W65O217(H2O)7]26–) are a well-established subclass of POMs [18–20]. The presence of a lone pair on the hetero element does not allow the closed Keggin unit to form. The dimeric, sandwich-type POMs based on two lone-pair-containing β-Keggin fragments (e.g., [β-AsIIIW9O33]9–, [β-SbIIIW9O33]9–) represent also a well-known class of sandwich-type POMs. The first members of this class ([M2(H2O)6(WO2)2(β-SbW9O33)2](14–2n)– (Mn+ = Fe3+, Co2+, Mn2+, Ni2+), were reported by Bösing et al. [21] in 1997. Since then some more isostructural derivatives have been characterized: ([M2(H2O)6(WO2)2(β-BiW9O33)2](14–2n)– (Mn+ = Fe3+, Co2+, Ni2+, Cu2+, Zn2+), [(VO(H2O)2)2(WO2)2(β-BiW9O33)2]10–, [Sn1.5(WO2(OH))0.5(WO2)2(β-XW9O33)2]10.5– (X = SbIII, BiIII), [M3(H2O)8(WO2)(β-TeW9O33)2]8– (M = Ni2+, Co2+), [(Zn(H2O)3)2(WO2)1.5(Zn(H2O)2)0.5(β-TeW9O33)2]8–, [(VO(H2O)2)1.5(WO(H2O)2)0.5(WO2)0.5(VO(H2O))1.5(β-TeW9O33)2]7– and [M4(H2O)10(β-XW9O33)2]n– (n = 6, X = AsIII and SbIII, M = FeIII and CrIII; n = 4, X = SeIV, TeIV, M = FeIII and CrIII; n = 8, X = SeIV, TeIV, M = MnII, CoII, NiII, ZnII, CdII and HgII) [22–26].
It becomes apparent that most of the known sandwich-type POMs contain divalent, paramagnetic transition metal ions. The motivation for synthesizing such compounds in the first place is usually centered around perceived applications in catalysis and medicine. Therefore, it is highly important to also investigate the solution properties of such POMs in addition to the solid state. The most sensitive and elegant tool for such studies is undoubtedly 183W NMR, but the paramagnetic nature of most sandwich-type POMs complicates matters. Therefore, it is of interest to prepare isostructural and diamagnetic analogs of these compounds.
In several cases it was possible to substitute the divalent, paramagnetic transition metal ions by diamagnetic Zn2+ and then 183W NMR was used to proof the structural integrity of the POMs in solution (e.g. [M3(H2O)3(α-XW9O33)2]12– (M = Cu2+, Zn2+; X = AsIII, SbIII) [15,27]. Obviously, substitution of iron(III)-containing POMs by zinc(II) results in derivatives with a different charge (e.g. [Fe4(H2O)10(β-SeW9O33)2]4– vs. [Zn4(H2O)10(β-SeW9O33)2]8–). In order to solve this problem, we decided to prepare indium(III)-substituted POMs with structures for which iron(III)-analogs exist.
Interestingly, very little work on indium substituted POMs has been reported to date [28–30]. In 1995, Meng et al. [28] described interaction of indium(III) with the trilacunary Keggin species α,β-[XW9O34]10– (X = Si, Ge) and based on 183W NMR they proposed trisubstituted, monomeric products. A few years later, Wasfi and Tribbitt [29] synthesized an indium(III)-substituted heteropolyfluorotungstate and they proposed a Dawson-like structure. Very recently, Limanski et al. [30] described the first structurally characterized, indium(III)-substituted polyanions.
Here we report on tri- and tetraindium(III)-substituted polyoxotungstates based on Keggin and Wells–Dawson fragments.
2 Experimental section
2.1 Synthesis
The lacunary precursors Na8H[B-α-PW9O34], Na12[P2W15O56]·24 H2O, Na9[α-AsW9O33] and Na9[α-SbW9O33] were synthesized according to published procedures and their purity was confirmed by infrared spectroscopy [21,31–33]. All other reagents were used as purchased without further purification.
2.1.1 d,l-(NH4)11[In3Cl2(B-α-PW9O34)2]·16H2O (1a)
A 0.39 g (1.76 mmol) sample of InCl3 was dissolved in 40 ml H2O followed by addition of 1.94 g (0.80 mmol) Na8H[B-α-PW9O34]. The pH of this solution was adjusted to 2 by addition of 4 M HCl and then it was heated to 80 °C for 1 h. After cooling to room temperature the solution was filtered. Then it was layered with 0.1 M NH4Cl and allowed to evaporate in an open beaker at room temperature. After 1–2 days a white crystalline product started to appear. Evaporation was allowed to continue until the solvent level had approached the solid product, which was filtered off and air-dried (1.6 g, yield 75%). IR: 1079(m), 1069(m), 1054(sh), 995(sh), 983(s), 963(sh), 885(m), 797(s), 728(sh), 681(sh), 595(w), 522(w) cm–1. Elemental analysis calc. (found): N 2.9 (2.7), In 6.4 (6.5), P 1.2 (1.1), W 61.7 (62.2), Cl 1.3 (1.0)%. 31P NMR (D2O, 293 K): δ –11.4 ppm; 183W NMR (D2O, 293 K): δ (relative intensities in parentheses) –98.3(1), –104.2(2), –128.6(2), –129.1(1), –133.1(2), –186.6(1) ppm.
2.1.2 d,l-(NH4)9Na8[In3Cl2(P2W15O56)2]·39 H2O (2a)
A 0.39 g (1.76 mmol) sample of InCl3 was dissolved in 40 ml H2O followed by addition of 3.52 g (0.80 mmol) Na12[P2W15O56]·24 H2O. The pH of this solution was adjusted to 2 by addition of 4 M HCl and then it was heated to 80 °C for 1 h. After cooling to room temperature the solution was filtered. Then it was layered with 0.1 M NH4Cl and allowed to evaporate in an open beaker at room temperature. After 1–2 days a white crystalline product started to appear. Evaporation was allowed to continue until the solvent level had approached the solid product, which was filtered off and air-dried (2.5 g, yield 69%). IR: 1092(s), 1059(m), 1017(w), 944(s), 917(s), 882(m), 831(sh), 810(sh), 764(s), 721(sh), 641(sh), 598(sh), 524(w) cm–1. Elemental analysis calc. (found): Na 2.1 (2.1), N 1.4 (1.2), In 3.9 (4.0), P 1.4 (1.3), W 62.0 (61.3), Cl 0.8 (0.9)%. 31P NMR (D2O, 293 K): δ (relative intensities in parenthesis) –7.1(1), –13.3(1) ppm.
2.1.3 RbNa3[In4(H2O)10(β-AsW9O32OH)2]·36 H2O (3a)
A 0.39 g (1.76 mmol) sample of InCl3 was dissolved in 40 ml H2O followed by addition of 2.00 g (0.80 mmol) Na9[α-AsW9O33]. The pH of this solution was adjusted to 2 by addition of 4 M HCl and then it was heated to 80 °C for 1 h. After cooling to room temperature the solution was filtered. Then it was layered with 0.1 M RbNO3 and allowed to evaporate in an open beaker at room temperature. After 1–2 days a white crystalline product started to appear. Evaporation was allowed to continue until the solvent level had approached the solid product, which was filtered off and air-dried. A total of 1.7 g (yield 72%) of crystalline product was obtained. IR: 955(m), 881(sh), 823(s), 794(s), 701(m), 614(sh), 510(sh), 478(w), 430(w) cm–1. Elemental analysis calcd. (found): Rb 1.4 (1.2), Na 1.2 (1.1), In 7.7 (7.5), As 2.5 (2.5), W 55.5 (55.0)%. 183W NMR (D2O, 293 K): δ (relative intensities in parenthesis) –102.3(1), –109.6(2), –153.7(2), –171.3(2), –199.2(2) ppm.
2.1.4 K4Na2[In4(H2O)10(β-SbW9O33)2]·30 H2O (4a)
The synthesis was identical to that of 1a, with the exception that 2.00 g (0.80 mmol) Na9[α-SbW9O33] was used instead of Na9[α-AsW9O33]. In addition, the solution was layered with 0.1 M KCl instead of RbNO3. In this case a total of 2.0 g (yield 82%) of crystalline product was obtained. IR: 949(m), 890(sh), 809(s), 693(m), 604(sh), 507(w), 467(w), 418(w) cm–1. Elemental analysis calc. (found): K 2.6 (2.8), Na 0.8 (0.5), In 7.7 (7.6), Sb 4.1 (4.0), W 55.2 (55.9)%.
We have also isolated the selenium(IV) and tellurium(IV) containing derivatives K4[In4(H2O)10(β-SeW9O33)2]·28 H2O (5a) and K4[In4(H2O)10(β-TeW9O33)2]·29 H2O (6a), respectively, as based on IR and elemental analysis. IR for 5a: 954(m), 890(sh), 819(s), 765(s), 653(m), 504(w), 447(w) cm–1. IR for 6a: 971(m), 889(m), 784(s), 749(m), 612(sh), 509(w), 423(w) cm–1. Elemental analysis for 5a calc. (found): K 2.7 (2.5), In 7.9 (7.6), Se 2.7 (2.8), W 56.8 (57.5)%. Elemental analysis for 6a calc. (found): K 2.6 (2.7), In 7.7 (7.5), Te 4.3 (4.1), W 55.7 (56.2)%.
Elemental analyses were performed by Kanti Labs Ltd. in Mississauga, Canada. IR spectra were recorded on a Nicolet Avatar FTIR spectrophotometer in a KBr pellet. All NMR spectra were recorded at room temperature with freshly synthesized solutions of 1–4 on a 400 MHz JEOL ECX instrument.
2.2 X-ray crystallography
Single-crystals of compounds 1a–4a were mounted on a glass fiber for indexing and intensity data collection at 173 K (1a, 4a) and 200 K (2a, 3a), respectively, on a Bruker D8 SMART APEX CCD single-crystal diffractometer using Mo Kα radiation (λ = 0.71073 Å). Direct methods were used to solve the structures and to locate the heavy atoms (SHELXS97). Then the remaining atoms were found from successive difference maps (SHELXL97). Routine Lorentz and polarization corrections were applied and an absorption correction was performed using the SADABS program [34]. Crystallographic data are summarized in Table 1.
Crystal data and structure refinement for (NH4)11[In3Cl2(PW9O34)2]·16 H2O (1a), (NH4)9Na8[In3Cl2(P2W15O56)2]·39 H2O (2a), RbNa3[In4(H2O)10(β-AsW9O32OH)2]·36 H2O (3a) and K4Na2[In4(H2O)10(β-SbW9O33)2]·30 H2O (4a)
| 1a | 2a | 3a | 4a | |
| Emperical formula | Cl2H76In3N11O84P2W18 | Cl2H114In3N9Na8O151P4W30 | As2H94In4Na3O112RbW18 | H80In4K4Na2O106Sb2W18 |
| Fw | 5361.5 | 8895.91 | 5959.8 | 5991.3 |
| Space group (number) | P21/n (14) | P | P | P |
| a (Å) | 17.5090(10) | 13.0643(5) | 12.8142(5) | 12.2306(9) |
| b (Å) | 12.7361(7) | 14.8901(6) | 12.8672(5) | 12.7622(10) |
| c (Å) | 18.7955(11) | 19.8603(8) | 16.1794(7) | 16.1639(12) |
| α (°) | 92.2920(10) | 91.1370(10) | 73.9890(10) | |
| β (°) | 107.3030(10) | 90.8680(10) | 105.9450(10) | 76.5550(10) |
| γ (°) | 100.5630(10) | 104.0980(10) | 86.2130(10) | |
| Volume (Å3) | 4001.6(4) | 3793.9(3) | 2477.02(17) | 2358.7(3) |
| Z | 2 | 1 | 1 | 1 |
| Temperature (°C) | –100 | –73.3 | –73.3 | –100 |
| Wavelength (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| dcalc (Mg m–3) | 4.404 | 3.851 | 3.918 | 4.167 |
| Absorbance coefficient (mm–1) | 26.837 | 23.301 | 22.995 | 23.674 |
| R [I > 2 σ(I)]a | 0.040 | 0.054 | 0.047 | 0.061 |
| Rw (all data)b | 0.075 | 0.133 | 0.119 | 0.113 |
a R = ∑||Fo|–|Fc||/∑|Fo|.
b Rw = [∑w(Fo2–Fc2)2/∑w(Fo2)2]1/2.
3 Results and discussion
The dimeric, chiral polyoxoanion [In3Cl2(B-α-PW9O34)2]11– (1) is composed of two (B-α-PW9O34) units linked via three In(III) ions (see Fig. 1). Polyanion 1 belongs to the well-known class of sandwich-type structures (Weakley type) and it represents the first indium-derivative [15]. Interestingly, only three indium centers are incorporated in 1 and it is very unusual that one of the inner positions is vacant. Both outer positions are occupied by indium ions with a terminal chloro ligand each. Therefore polyanion 1 exhibits C2 point group symmetry (as opposed to C2h for the tetrasubstituted transition metal analogs, e.g. [Mn4(H2O)2(B-α-PW9O34)2]10–) indicating that it is chiral. The only other trisubstituted analog of 1 known to date is [Ni3Na(H2O)2(B-α-PW9O34)2]11–, but in this case the ‘vacant’ site is in an outer position and it is occupied by a sodium ion in the solid state [17]. Bond lengths and angles associated with the central indium-oxo fragment of 1 are shown in Table 2.
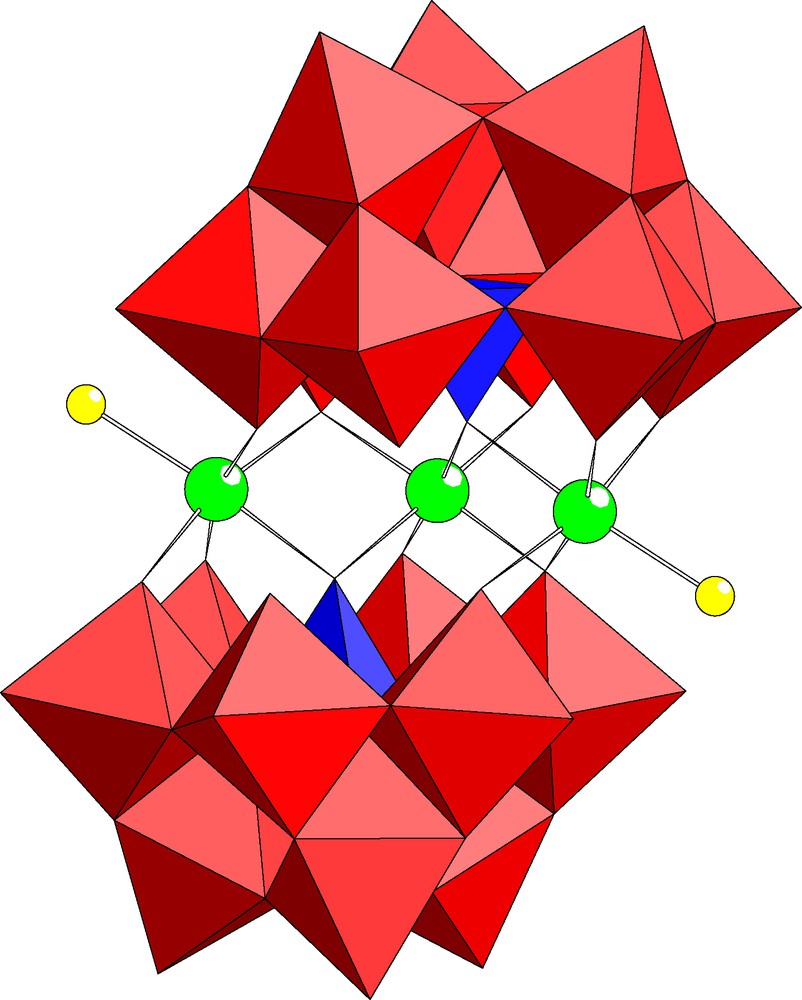
Combined polyhedral/ball and stick representation of [In3Cl2(PW9O34)2]11– (1). The red octahedra represent WO6, the blue tetrahedra represent PO4 and the balls represent indium (green) and chlorine (yellow).
Selected bond lengths and angles in [In3Cl2(PW9O34)2]11– (1), [In3Cl2(P2W15O56)2]17– (2)
| 1 | 2 | ||
| In1–O4I1 | 2.061(8) | In1–O10I | 2.087(9) |
| In1–O7A | 2.082(8) | In1–O13A | 2.115(9) |
| In1–O5IN | 2.133(7) | In1–O15I | 2.136(9) |
| In1–O6I2 | 2.146(7) | In1–O14A | 2.165(9) |
| In1–O4P | 2.320(8) | In1–O4P2 | 2.248(8) |
| In1–O4P | 2.323(7) | In1–O4P2 | 2.274(8) |
| In2–O8A | 2.104(7) | In2–O11I | 2.120(9) |
| In2–O9A | 2.118(7) | In2–O14A | 2.126(8) |
| In2–O6I2 | 2.139(7) | In2–O12I | 2.126(9) |
| In2–O5IN | 2.147(7) | In2–O15I | 2.134(8) |
| In2–O4P | 2.287(7) | In2–O4P2 | 2.224(9) |
| In2–Cl1 | 2.418(3) | In2–Cl1 | 2.394(6) |
| O4I1–In1–O7A | 95.7(3) | O10I–In1–O13A | 94.5(4) |
| O4I1–In1–O5IN | 101.7(3) | O10I–In1–O15I | 98.6(3) |
| O7A–In1–O5IN | 87.6(3) | O13A–In1–O15I | 87.3(3) |
| O4I1–In1–O6I2 | 86.1(3) | O10I–In1–O14A | 87.6(3) |
| O7A–In1–O6I2 | 100.2(3) | O13A–In1–O14A | 99.7(3) |
| O5IN–In1–O6I2 | 168.5(3) | O15I–In1–O14A | 170.3(3) |
| O4I1–In1–O4P | 175.8(3) | O10I–In1–O4P2 | 175.2(3) |
| O7A–In1–O4P | 88.1(3) | O13A–In1–O4P2 | 90.3(3) |
| O5IN–In1–O4P | 80.3(3) | O15I–In1–O4P2 | 81.9(3) |
| O6I2–In1–O4P | 91.4(3) | O14A–In1–O4P2 | 91.3(3) |
| O4I1–In1–O4P | 88.1(3) | O10I–In1–O4P2 | 88.9(3) |
| O7A–In1–O4P | 176.2(3) | O13A–In1–O4P2 | 176.5(3) |
| O5IN–In1–O4P | 91.9(3) | O15I–In1–O4P2 | 91.3(3) |
| O6I2–In1–O4P | 79.7(3) | O14A–In1–O4P2 | 81.4(3) |
| O4P–In1–O4P | 88.1(3) | O4P2–In1–O4P2 | 86.3(3) |
| O8A–In2–O9A | 94.4(3) | O11I–In2–O14A | 89.8(3) |
| O8A–In2–O6I2 | 165.3(3) | O11I–In2–O12I | 91.6(3) |
| O9A–In2–O6I2 | 88.7(3) | O14A–In2–O12I | 169.7(3) |
| O8A–In2–O5IN | 88.7(3) | O11I–In2–O15I | 169.5(3) |
| O9A–In2–O5IN | 167.4(3) | O14A–In2–O15I | 87.2(3) |
| O6I2–In2–O5IN | 85.4(3) | O12I–In2–O15I | 89.6(3) |
| O8A–In2–O4P | 85.1(3) | O11I–In2–O4P2 | 87.1(3) |
| O9A–In2–O4P | 87.3(3) | O14A–In2–O4P2 | 83.4(3) |
| O6I2–In2–O4P | 80.7(3) | O12I–In2–O4P2 | 86.5(3) |
| O5IN–In2–O4P | 80.8(3) | O15I–In2–O4P2 | 82.6(3) |
| O8A–In2–Cl1 | 92.4(2) | O11I–In2–Cl1 | 89.0(3) |
| O9A–In2–Cl1 | 89.30(19) | O14A–In2–Cl1 | 98.7(3) |
| O6I2–In2–Cl1 | 101.96(19) | O12I–In2–Cl1 | 91.5(3) |
| O5IN–In2–Cl1 | 102.80(19) | O15I–In2–Cl1 | 101.4(3) |
| O4P–In2–Cl1 | 175.6(2) | O4P2–In2–Cl1 | 175.5(3) |
The structure of 1 and its chiral nature somewhat resemble the Tourné et al. [35] polyanion [WM3(H2O)2(B-α-XW9O34)2]12– (X = M = Zn2+, Co2+). Due to the fact that most polyoxoanions are not inherently chiral, Tourné’s species and especially its noble metal containing derivatives (e.g. [WZnRuIII2(H2O)2(B-α-ZnW9O34)2]10–) have been studied intensely for their potentially asymmetric catalytic activity [36]. It remains to be seen if chiral, transition metal substituted derivatives of 1 can be prepared.
Polyanion 1 was synthsesized by interaction of In3+ ions with the trilacunary precursor [B-α-PW9O34]9– in aqueous, acidic medium. We discovered that the trisubstituted 1 is obtained even in the presence of excess In3+ ions. Additional studies are needed in order to explain why the tri- rather than the tetrasubstituted product is formed and also, why the vacancy is in an interior site.
As polyanion 1 is diamagnetic we performed 183W and 31P NMR studies in solution. The 31P NMR spectrum of 1 exhibits one signal at –11.4 ppm and the 183W NMR spectrum shows six peaks at –98.3(1), –104.2(2), –128.6(2), –129.1(1), –133.1(2) and –186.6(1) ppm, respectively (see Fig. 2). Initially this six-line pattern with intensity ratios 2:2:2:1:1:1 may seem unexpected, but it can be rationalized as follows. The hypothetical, tetrasubstitued derivative [In4Cl2(B-α-PW9O34)2]8– has C2h symmetry and would therefore be expected to show five lines in 183W NMR with intensity ratios 2:2:2:2:1. Indeed, we have obtained exactly this pattern for the tetrasubstituted, isostructural Zn2+ and Cd2+ derivatives [M4(H2O)2(B-α-GeW9O34)2]12– (M = Zn2+, Cd2+) [15]. Removal of one of the inner indium atoms from [In4Cl2(B-α-PW9O34)2]8– results in 1 with C2 point group symmetry. Consequently, nine peaks are expected in 183W NMR. However, close inspection of the structure of 1 (see Fig. 1) indicates that the three tungsten atoms in the Keggin cap and four of the six tungsten atoms in the Keggin belt are not likely to be affected significantly by removal of an inner indium atom as they do not share oxo ligands with that indium atom. The inner indium atoms are coordinated to only three oxo groups of each Keggin half-unit, involving two belt tungsten atoms (W6, W7) and the phosphorus hetero atom. The six-line 183W NMR spectrum of 1 with intensity ratios 2:2:2:1:1:1 indicates that only one of these two belt tungsten atoms is significantly affected by removal of an inner indium atom. Clearly, this must be W7 which experiences a major change in its coordination sphere as a result of In atom removal. Before, it has only one terminal oxo ligand and after it has two terminal, cis-related oxo ligands.

Tungsten-183 NMR spectrum of [In3Cl2(B-α-PW9O34)2]11– (1) at 293 K.
The dimeric polyanion [In3Cl2(P2W15O56)2]17– (2) represents the Wells–Dawson analog of 1 (see Fig. 3). Polyanion 2 contains three indium centers sandwiched in between two trilacunary (P2W15O56) units and one of the inner positions is vacant. In analogy to 1 the terminal ligands of the outer indium atoms in 2 are also chloride ions. As a result, polyanion 2 exhibits also C2 point group symmetry and is therefore chiral. The bond lengths and angles associated with the central indium-oxo fragment of 2 are shown in Table 2. Some other trisubstituted analogs of 2 exist, but in all cases the ‘vacant’ site is in an outer position and it is occupied by a sodium ion in the solid state (e.g., [Fe2(FeOH2)(NaOH2)(P2W15O56)2]14–) [16].
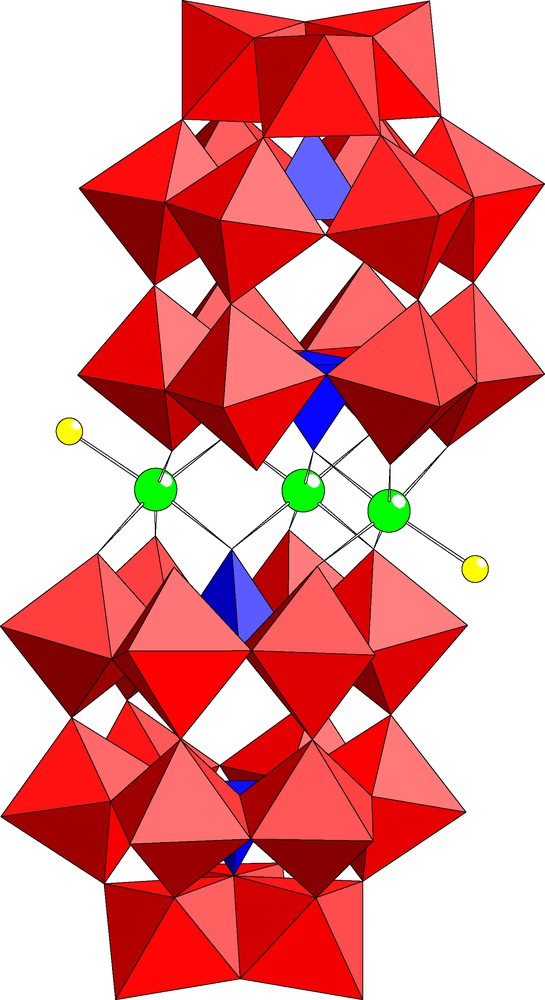
Combined polyhedral/ball and stick representation of [In3Cl2(P2W15O56)2]17– (2). The color code is the same as in Fig. 1.
Polyanion 2 was synthesized by interaction of In3+ ions with the trilacunary precursor [P2W15O56]12– in aqueous, acidic medium. We discovered that the trisubstituted 2 is formed even in the presence of excess In3+ ions. As polyanion 2 is diamagnetic we performed 183W and 31P NMR studies in solution. The latter exhibits two signals of equal intensity at –7.1 and –13.3 ppm, respectively, which is in agreement with the solid state structure. The two Wells–Dawson fragments in 2 are equivalent and they contain two P-atoms each. The signal at –7.1 ppm can be assigned to the P-atom closer to the indium-core, whereas the signal at –13.3 ppm corresponds to the more distant P-atom. The 183W NMR spectrum of 2 is expected to show around 9–10 peaks (based on the arguments used above for 1) and we see this number of signals between –80 and –240 ppm. However, the poor signal-to-noise ratio of our spectrum did not allow us to identify all peaks unequivocally.
Although polyanions 1 and 2 are chiral they have crystallized as a racemic mixture. Our crystallographic studies have revealed that both inner indium positions are occupied, but only with occupancy factors of 0.5 each. This means that individual molecules of 1 and 2 contain only one indium atom in one of the two possible inner positions. As the synthesis procedure of 1 and 2 is not stereoselective, both enantiomers are formed in equal amounts. Apparently the crystallization process is also not stereoselective, which is not a surprise as the chiral site in 1 and 2 is somewhat hidden by the two Keggin caps and as a result is not expected to influence polyanion packing significantly. Therefore, both enantiomers orient randomly within the solid state lattices of 1a and 2a leading to the crystallographic observations described above.
The indium substituted, lone pair containing polyoxoanions [In4(H2O)10(β-AsW9O32OH)2]4– (3) and [In4(H2O)10(β-SbW9O33)2]6– (4) are isostructural. They consist of two β-XW9 (X = AsIII, SbIII) Keggin moieties linked by four In3+ ions resulting in a structure with idealized C2h symmetry (see Fig. 4). The four In3+ ions consist of two inequivalent pairs, the inner two In3+ ions have two terminal H2O ligands and the outer two In3+ ions have three terminal H2O ligands. Polyanions 3 and 4 were synthesized in aqueous acidic medium (pH 2) from interaction of In3+ ions with the lacunary Keggin precursors [α-AsW9O33]9– and [α-SbW9O33]9–, respectively. Therefore the mechanism of formation of 3 and 4 involves insertion, isomerization (α → β) and dimerization. The bond lengths and angles associated with the central indium-oxo fragments of 3 and 4 are shown in Table 3.

Combined polyhedral/ball and stick representation representative of [In4(H2O)10(β-AsW9O32OH)2]4– (3) and [In4(H2O)10(β-SbW9O33)2]6– (4). The WO6 octahedra are shown in red and the balls represent indium (green), arsenic/antimony (blue) and water molecules (red).
Selected bond lengths and angles in [In4(H2O)10(β-AsW9O32OH)2]4– (3) and [In4(H2O)10(β-SbW9O33)2]6– (4)
| 3 | 4 | ||
| In(1)–O(3I1) | 2.069(8) | In(1)–O(3IN) | 2.083(11) |
| In(1)–O(7I1) | 2.073(7) | In(1)–O(6IN) | 2.109(10) |
| In(1)–O(6T)#1 | 2.107(7) | In(1)–O(9B)#2 | 2.120(10) |
| In(1)–O(6I1) | 2.109(7) | In(1)–O(1I1) | 2.146(10) |
| In(1)–O(2WI) | 2.192(8) | In(1)–O(3I1) | 2.164(12) |
| In(1)–O(1WI) | 2.206(8) | In(1)–O(2I1) | 2.193(12) |
| In(2)–O(4I2) | 2.084(7) | In(2)–O(1A)#2 | 2.059(10) |
| In(2)–O(5I2) | 2.112(7) | In(2)–O(5IN) | 2.072(11) |
| In(2)–O(4WI) | 2.153(8) | In(2)–O(9IN) | 2.110(11) |
| In(2)–O(5WI) | 2.156(8) | In(2)–O(9A)#2 | 2.117(10) |
| In(2)–O(6I2) | 2.160(8) | In(2)–O(2I2) | 2.170(12) |
| In(2)–O(3WI) | 2.206(9) | In(2)–O(1I2) | 2.174(11) |
| As(1)–O(3AS) | 1.794(7) | Sb(1)–O(2SB) | 1.989(9) |
| As(1)–O(1AS) | 1.810(7) | Sb(1)–O(1SB) | 2.014(10) |
| As(1)–O(2AS) | 1.814(7) | Sb(1)–O(3SB) | 2.014(10) |
| O(3I1)–In(1)–O(7I1) | 173.8(3) | O(3IN)–In(1)–O(6IN) | 86.1(4) |
| O(3I1)–In(1)–O(6T)#1 | 89.4(3) | O(3IN)–In(1)–O(9B)#2 | 99.4(4) |
| O(7I1)–In(1)–O(6T)#1 | 95.0(3) | O(6IN)–In(1)–O(9B)#2 | 95.6(4) |
| O(3I1)–In(1)–O(6I1) | 92.3(3) | O(3IN)–In(1)–O(1I1) | 95.3(4) |
| O(7I1)–In(1)–O(6I1) | 91.9(3) | O(6IN)–In(1)–O(1I1) | 177.1(4) |
| O(6T)#1–In(1)–O(6I1) | 92.4(3) | O(9B)#2–In(1)–O(1I1) | 86.7(4) |
| O(3I1)–In(1)–O(2WI) | 84.7(3) | O(3IN)–In(1)–O(3I1) | 174.4(4) |
| O(7I1)–In(1)–O(2WI) | 90.4(3) | O(6IN)–In(1)–O(3I1) | 93.7(4) |
| O(6T)#1–In(1)–O(2WI) | 172.0(3) | O(9B)#2–In(1)–O(3I1) | 86.2(4) |
| O(6I1)–In(1)–O(2WI) | 93.2(3) | O(1I1)–In(1)–O(3I1) | 84.6(4) |
| O(3I1)–In(1)–O(1WI) | 90.4(3) | O(3IN)–In(1)–O(2I1) | 91.9(4) |
| O(7I1)–In(1)–O(1WI) | 85.5(3) | O(6IN)–In(1)–O(2I1) | 93.2(4) |
| O(6T)#1–In(1)–O(1WI) | 86.8(3) | O(9B)#2–In(1)–O(2I1) | 166.1(4) |
| O(6I1)–In(1)–O(1WI) | 177.2(3) | O(1I1)–In(1)–O(2I1) | 84.2(4) |
| O(2WI)–In(1)–O(1WI) | 87.9(3) | O(3I1)–In(1)–O(2I1) | 82.5(5) |
| O(4I2)–In(2)–O(5I2) | 90.9(3) | O(1A)#2–In(2)–O(5IN) | 177.4(4) |
| O(4I2)–In(2)–O(4WI) | 91.6(3) | O(1A)#2–In(2)–O(9IN) | 92.2(4) |
| O(5I2)–In(2)–O(4WI) | 174.6(3) | O(5IN)–In(2)–O(9IN) | 90.0(4) |
| O(4I2)–In(2)–O(5WI) | 175.2(3) | O(1A)#2–In(2)–O(9A)#2 | 89.6(4) |
| O(5I2)–In(2)–O(5WI) | 90.0(3) | O(5IN)–In(2)–O(9A)#2 | 89.0(4) |
| O(4WI)–In(2)–O(5WI) | 87.2(3) | O(9IN)–In(2)–O(9A)#2 | 88.8(4) |
| O(4I2)–In(2)–O(6I2) | 96.0(3) | O(1A)#2–In(2)–O(2I2) | 94.3(4) |
| O(5I2)–In(2)–O(6I2) | 98.8(3) | O(5IN)–In(2)–O(2I2) | 83.5(4) |
| O(4WI)–In(2)–O(6I2) | 85.7(3) | O(9IN)–In(2)–O(2I2) | 173.5(4) |
| O(5WI)–In(2)–O(6I2) | 88.5(3) | O(9A)#2–In(2)–O(2I2) | 90.3(4) |
| O(4I2)–In(2)–O(3WI) | 93.3(3) | O(1A)#2–In(2)–O(1I2) | 88.1(4) |
| O(5I2)–In(2)–O(3WI) | 90.9(3) | O(5IN)–In(2)–O(1I2) | 93.2(4) |
| O(4WI)–In(2)–O(3WI) | 84.1(3) | O(9IN)–In(2)–O(1I2) | 94.4(4) |
| O(5WI)–In(2)–O(3WI) | 82.0(3) | O(9A)#2–In(2)–O(1I2) | 176.2(4) |
| O(6I2)–In(2)–O(3WI) | 166.4(3) | O(2I2)–In(2)–O(1I2) | 86.8(4) |
| O(3AS)–As(1)–O(1AS) | 98.9(3) | O(2SB)–Sb(1)–O(1SB) | 90.7(4) |
| O(3AS)–As(1)–O(2AS) | 96.8(3) | O(2SB)–Sb(1)–O(3SB) | 93.1(4) |
| O(1AS)–As(1)–O(2AS) | 98.1(3) | O(1SB)–Sb(1)–O(3SB) | 93.3(4) |
Tungsten-183 NMR on freshly synthesized solutions of 3 resulted in five peaks (relative intensities in parenthesis) at –102.3(1), –109.6(2), –153.7(2), –171.3(2) and –199.2(2) ppm, respectively (see Fig. 5). This result indicates that in solution a species with C2h symmetry is present, which is in complete agreement with the solid state structure of 3. Furthermore, the NMR spectrum does not change even after several weeks, indicating the high stability of 3 in aqueous solution. The 183W NMR behavior of the antimony derivative 4 is apparently more complex and requires additional studies. Our preliminary results on freshly synthesized solutions of 4 resulted in six peaks at –80.9, –99.5, –117.5, –133.9, –160.6 and –176.7 ppm.

Tungsten-183 NMR spectrum of [In4(H2O)10(β-AsW9O32OH)2]4– (3) at 293 K.
Very recently (while this manuscript was in preparation), Limanski et al. [30] reported on the solid-state structures of the closely related compounds Na5nH2n[(In(H2O)2)1.5(Na(H2O)2)0.5(In(H2O)2)2(SbW9O33)2]n·28nH2O and Na2K2H2[(In(H2O)3)2(In(H2O)2)2(AsW9O33)2]·37H2O. Although the solid-state structures of Krebs’ compounds and ours are different, the latter contains a polyanion identical to 3. On the other hand, the antimony(III)-containing polyanion of Krebs et al. is different from 4 as the outer indium positions are partially (25%) occupied by sodium ions resulting in a polymeric solid state structure.
Bond valence sum (BVS) calculations on 3 and 4 resulted in the conclusion that all terminal ligands of the indium atoms are water molecules [37]. For 4 we could not identify any other protonation sites, but for 3 we noticed that two In–O–W bridging oxygens (O5I2, O5I2′) are actually hydroxo groups. This results in a charge of –4 for 3 and –6 for 4, which is somewhat surprising as both species were synthesized at exactly the same pH.
It is of interest to compare the synthetic conditions for the preparation of Krebs’ and our compounds. Besides using a higher concentration of the reagents (by about a factor of 2), Krebs and coworkers also performed their syntheses at pH 6.5–7, whereas we worked in much more acidic medium (pH 2). We used such acidic conditions because former work of Krebs et al. and also our own has demonstrated that [α-XW9O33]9– and [β-XW9O33]9– (X = AsIII, SbIII) are in equilibrium in aqueous solution [20,21,24,27]. The former dominates in neutral medium whereas the latter is present in acidic solution. Our synthetic conditions of 3 and 4 are in full agreement with this, as both species are formed easily in acidic medium. The recent results of Krebs et al. indicate that 3 is also stable in the neutral pH range, but this is apparently not the case for 4. This observation combined with the different degree of protonation for 3 and 4 deserves attention and is further supported by our 183W NMR studies (see above).
We have already observed previously that the products resulting from interaction of transition metal ions with [α-AsW9O33]9– and [β-SbW9O33]9– are not solely determined by the pH, but also by the type of transition metal ion and its preferred coordination geometry. For example, we were able to synthesize the Fe3+ analogues of 3 and 4, but not the Cu2+ and Zn2+ derivatives [24,27]. Reaction of the latter metal ions with [α-XW9O33]9– (X = AsIII, SbIII) resulted in the polyanions [M3(H2O)3(α-XW9O33)2]12– (M = Cu2+, Zn2+; X = AsIII, SbIII), no matter what the pH. The Cu2+ and Zn2+ ions have a square pyramidal coordination geometry in this structure, whereas the coordination geometry of all four In3+ ions in 3 and 4 is octahedral. Interestingly, Cr3+ was the only other first-row transition metal ion besides Fe3+ for which we could synthesize the tetra-substituted structure with AsIII and SbIII as hetero atoms [24]. It becomes apparent, that all three ions which allow formation of this structural type are trivalent (In3+, Fe3+, Cr3+). However, by using SeIV and TeIV as hetero atoms we could also incorporate divalent transiton metal ions (e.g. Mn2+, Co2+, Ni2+, Zn2+, Cd2+, Hg2+) [24]. These results indicate that the tetra-substituted Krebs-type structure is most stable within the charge limits of –4 and –8.
The differences of 3 and 4 in their solution and solid state properties (e.g. protonation, charge, pH-dependent stability) led us to check if the different hetero atoms (AsIII, SbIII) perhaps initiate small structural changes (e.g., bond lengths, angles) in the polyanions. As expected, the As–O bond lengths are shorter than the Sb–O bonds (see Table 2) and the observed hetero atom separations X···X within 3 (As···As = 6.26 Å) and 4 (Sb···Sb = 5.87 Å) are fully consistent with this. On the other hand, the type of hetero group has no significant effect on the W–O and In–O bond lengths in 3 and 4 (see Table 2). Therefore we conclude that there are no obvious structural differences in 3 and 4. Furthermore, there is no significant lone-pair/lone-pair repulsion involving the lone pairs of the two hetero atoms. Both conclusions are fully consistent with our observations on the iron(III)-substituted derivatives of 3 and 4 [24]. In fact, the hetero atom separations in 3 and 4 (see above) are even larger than in [Fe4(H2O)10(β-AsW9O33)2]6– (As···As = 6.03 Å) and in [Fe4(H2O)10(β-SbW9O33)2]6– (Sb···Sb = 5.68 Å), which reflects the larger In–O bond lengths compared to Fe–O (the ionic radii of In3+ and Fe3+ are 0.94 and 0.79 Å, respectively).
4 Conclusions
We have synthesized and structurally characterized four novel indium(III)-substituted, sandwich-type polyoxoanions. The tri-indium substituted tungstophosphates [In3Cl2(B-α-PW9O34)2]11– (1) and [In3Cl2(P2W15O56)2]17– (2) belong to the class of Weakley-type sandwich polyanions and they represent the first indium-containing derivatives. Polyanions 1 and 2 are also stable in solution as indicated by a one-line (1) and a two-line (2) 31P NMR pattern. The most important structural feature of 1 and 2 is the fact that they are chiral (C2 symmetry), which results from a vacancy in one of the inner indium positions. Therefore 1 and 2 are of interest for catalytic and medicinal applications. Currently we investigate if chiral, transition metal substituted derivatives of 1 and 2 can be formed.
The lone pair containing, tetra-indium substituted polyoxotungstates [In4(H2O)10(β-AsW9O32OH)2]4– (3) and [In4(H2O)10(β-SbW9O33)2]6– (4) belong to the class of Krebs-type sandwich structures. Polyanions 3 and 4 consist of two B-β-(XW9O33) (X = AsIII, SbIII) Keggin moieties linked via four octahedral indium atoms leading to a dimeric structure with nominal C2h symmetry. Polyanion 3 is also stable in solution as indicated by a five line pattern (1:2:2:2:2) in 183W NMR. We have also synthesized the Se(IV) and Te(IV) analogues of 3 and 4.
5 Supplementary material
Four X-ray crystallographic files in CIF format. This material can be obtained from the Fachinformationszentrum Karlsruhe, Abt. PROKA, 76344 Eggenstein-Leopoldshafen, Germany (fax: +49-7247-808-666; e-mail: crysdata@fiz-karlsruhe.de) on quoting the depository numbers CSD-414099, 414101, 414280 and 414281, respectively.
Acknowledgements
U.K. thanks the International University Bremen for research support. U.K. also highly appreciates that the Florida State University Chemistry Department (USA) allowed him unlimited access to the single-crystal X-ray diffractometer. Figs. 1,3 and 4 were generated by Diamond Version 2.1e (copyright Crystal Impact GbR).