

The use of pyridine derivatives has shown an important development, particularly in the field of materials and functionalisation of surfaces. [1] Many applications such as photoswitch sensors for transition metals [1a,b], supramolecular self-assembly of multilayers organometallic thin films [1c,d] for photocurrent-generating systems [1e] have been described. The anchoring of the pyridine moiety to prepare self-assembled monolayers (SAMs) is usually achieved by reacting thiol groups on gold [1a–e] or catechols [2a] and carboxylic [2b] or phosphonic acids [2c,d] at the surface of metal oxides. In the course of our program concerning the functionalisation of metal oxide nanoparticles with phosphonate derivatives of pyridine [2d], we present here the synthesis of ethyl and di-tert-butyl phosphonates π-conjugated of pyridine and the studies of their reactivity and complexation with boron and ruthenium derivatives.
Stilbazoles and π-conjugated derivatives of pyridines have been synthesized by using the Wittig–Horner [3a] or Heck [3b] reactions. A drawback of the Horner reaction is that a mixture of cis–trans isomers could be obtained. Trans isomer is preferred because of better coplanarity of the conjugated system and enhanced electronic effects [4]. We thus turned to the Heck [5] reaction and first prepared stilbazole derivatives (Scheme 1).

Synthesis of stilbazole derivatives. Reagents and conditions: i) 4% Pd(OAc)2, 15% TOP, Et3N, toluene, 115 °C.
The Heck reaction was performed using standard homogeneous conditions with 4-vinylpyridine in the presence of the bromophosphonates 1a–d that were prepared as described in literature procedures [6]. The reaction led efficiently to trans compounds 2a-d in good yields.
We also prepared more conjugated derivatives by using two successive Heck reactions starting from para-iodobromobenzene and 4-vinylpyridine, then cross-coupling intermediate 3 with diethyl and di-tert-butyl 4-vinylbenzylphosphonates (Scheme 2).
Synthesis of stilbazole derivatives. Reagents and conditions: i) 4% Pd(OAc)2, 15% triorthotolyl-phosphine (TOP), Et3N, toluene, 115 °C.
The products 4a and 4b were obtained in 20% and 41% overall yield respectively. The yield was lower with the more emcumbered di-tert-butyl phosphonate ester. The diethyl and di-tert-butyl phosphonates were hydrolyzed with TMSBr and HCl 1 N, respectively. The two methods efficiently led to the acids (in the zwitterionic form) in good yields (Scheme 3).
Hydrolysis of phosphonates esters. Reagents and conditions: i) a) 3.5 equiv TMSBr, CH2Cl2, 12 h; b) H2O. ii) HCl 1N, 12 h, RT.
Stilbazolium, π-conjugated pyridinium salts and borane adducts of π-conjugated derivatives of pyridine are known to give strong non-linear optical properties [7]. We thus first reacted compounds 2a, 2c, 4b, 2d with methyl iodide (Scheme 4) in order to study the reactivity of the molecules (regiochemistry) and to obtain the required π-conjugated pyridinium salt.

Methylation of pyridine derivatives. Reagents and conditions: i) 1.5 equiv MeI, MeCN, RT, 12 h.
The reaction was very efficient, leading to salts 6a–d, in excellent yield and the phosphonate moiety did not interfere. Complexation of derivatives 2a, 2b, 5d, with BF3.Et2O was then performed (Scheme 5). In all the cases the reaction was regioselective giving adducts with the nitrogen of the pyridine moiety, no reaction was observed with the P=O function of esters 2a, 2b or acid 5d, even by addition of 2 equivalents of BF3.
Complexation of pyridine derivatives. Reagents and conditions: i) 1 equiv BF3Et2O, THF, RT, 4 h.
No cleavage of the sensitive t-Bu ester of compound 2b occurred. Indeed, 11B MAS solid-state NMR of products 7b and 7d showed only one signal at –1.9 and –0.9 ppm respectively, and no modification of the 31P NMR signal was observed after complexation. The trifluoroborate–pyridine adduct was very stable and was not cleaved during the hydrolysis of the phosphonate ester moiety of compound 7a. Acid 7e was obtained in good yield (Scheme 3).
Properties of organometallic complexes of pyridine have been widely studied [8] in fields such as supramolecular assemblies[8a,b], design of sensors [8c], catalysis [8d]. However, tetrakis pyridine ruthenium complexes [9] have been much less described than their bi or ter pyridine counterparts, as the former complexes are less stable. The ligands already described are pyridine[9a–c], 4-vinyl-pyridine [9d], 4-formylpyridine[9e], 4-acetylpyridine[9f], 4-methylpyridine[9g]. We decided to check the complexation of π-conjugated molecules 3, 2a, 4b (Scheme 6), starting from RuCl3 in refluxing EtOH, H2O [9d].
Synthesis of Ru complexes. Reagents and conditions: i) EtOH, H2O, reflux.
The complexes 8 were analysed by 1H and 13C NMR, and mass spectrometry (Table 1).
NMR and Mass data for ruthenium complexes 8
| Complexes | Mass Fab+ |
|
|
| 8a | 1210 (MH+), 950, 691, 260 |
| 157.6 |
| 8b | 1497 (MH+), 1167, 835, 332 |
| 158.1 |
| 8c | 1905 (MH+), 1473, 434 |
| 158.1 |
1H NMR showed only one signal for the α proton of the complexes 8a–c, in agreement with a trans geometry of the complexes (a cis geometry would have given two signals). 13C NMR showed a downfield-shielded signal at 158 ppm for the α carbons of compounds 8a–c compared with the precursors whose α carbons presented a signal at 150 ppm. This shield confirmed the complexation. Mass spectra showed the molecular peak for all the complexes with fragments corresponding to the loss of 1 to 3 ligands. Thus the presence of vinylene–phenylene groups did not perturb the complexation of the pyridine moiety with Ru and the complexes were obtained in excellent yield. Data concerning optical properties of selected π-conjugated pyridine compounds 2a, 2c, 4a are listed in Table 2.
Optical data for compounds 2a, 2c, 4a, (THF 10–4 M)
| Compound |
| Fluorescence | |
| Ex (nm) | Em λmax (nm) | ||
| 2a | 311 (3090) | 280 | 345 |
| 2c | 322 (18500) | 290 | 355 |
| 4a | 356 (45700) | 300 | 437 |
All the spectra were recorded at 10–4M in THF. The UV characteristic π−π* band of the derivatives and the fluorescence λmax emission are both red-shifted by increasing conjugation. After methylation or complexation with BF3, the absorption and emission spectra of compounds 6a and 7a were very similar (Figs. 1 and 2). Comparing with precursor 2a, an important bathochromic shift to 351 nm was observed for the π−π* band of the two compounds. These shifts could be explained by a quaternarization of the nitrogen and hyperconjugation effect with the methyl group [10], and by an electronic delocalisation on the boron atom [7], respectively. The λmax fluorescence emission was thus observed at 435 and 432 nm, respectively. With the ruthenium complex 8b, the absorption spectrum was different from that of compounds 6a and 7a (Fig. 3). It presented the π−π* band at 285 nm and a new band at 448 nm that characterizes the metal–ligand charge transfer [8d]. We did not observe significant fluorescence of compound 8b compared with 6a and 7a. Indeed, ruthenium–bpyridine complexes with trans halogen ligands are known to display practically no emission at RT due to very short-lived excited states, as these excited states are rapidly desactivated by non-radiative processes [11].
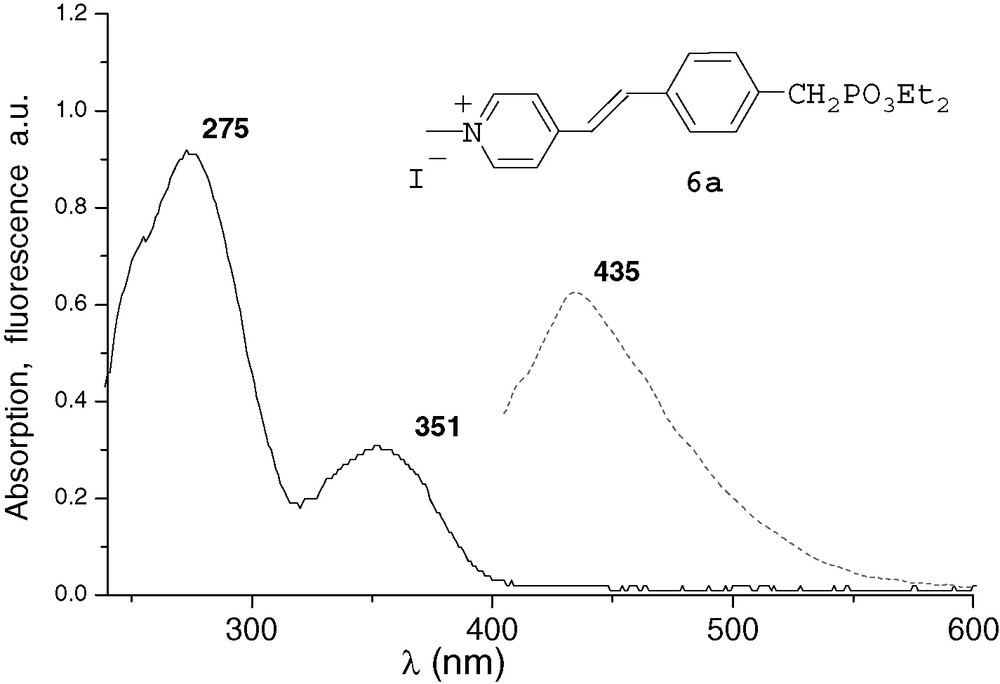
UV and fluorescence (ex 330 nm) of compound 6a.
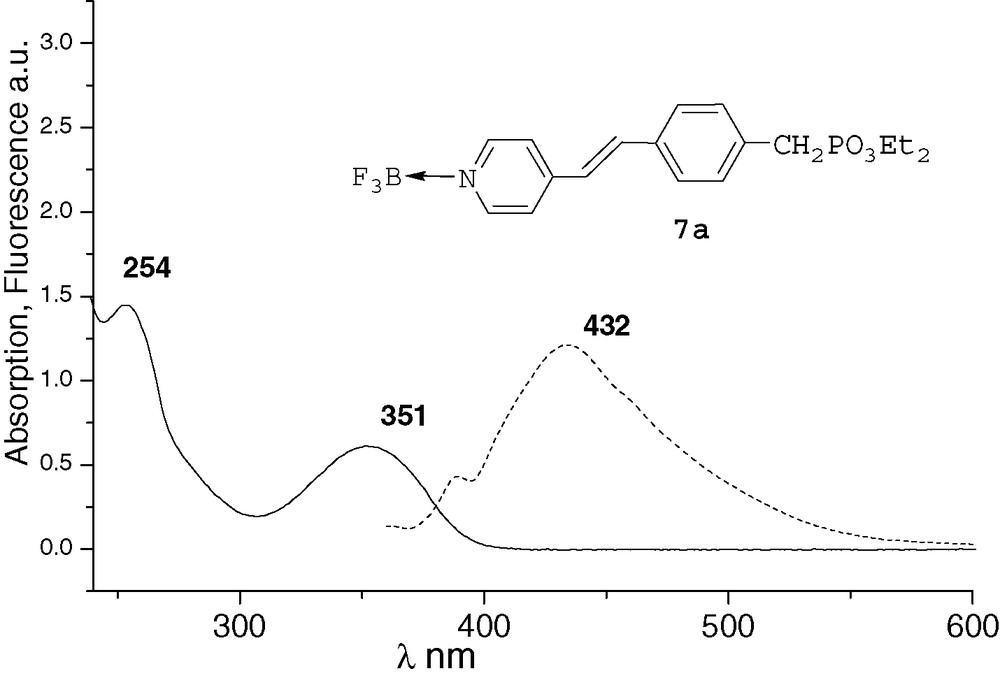
UV and fluorescence (ex 330 nm) of compound 7a.
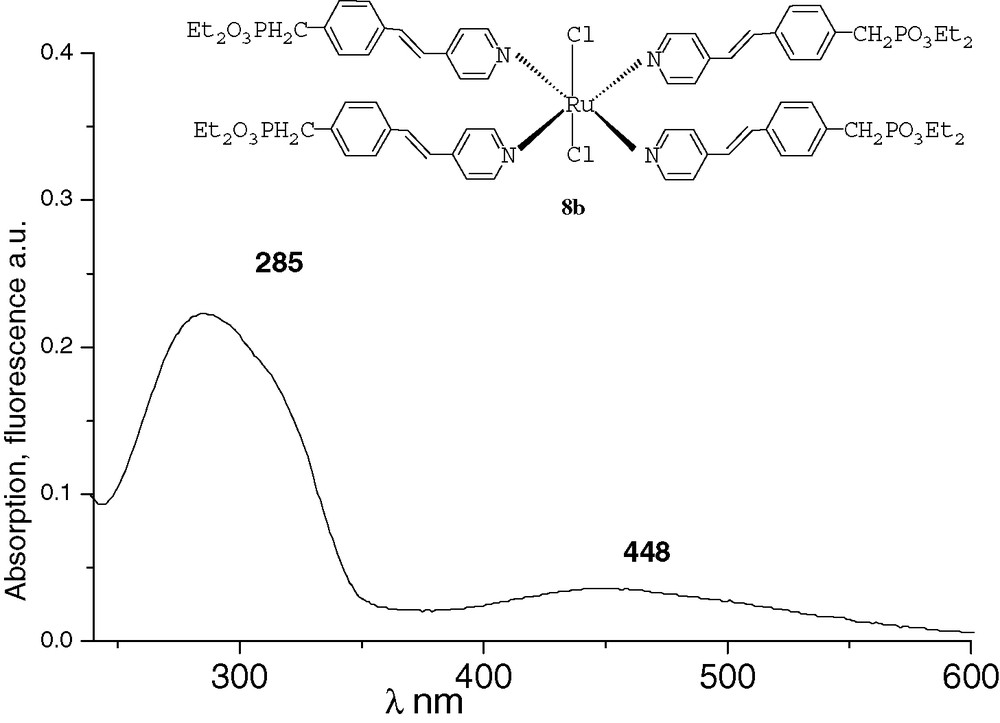
UV of complex 8b.
In conclusion, the Heck reaction was very efficient for the synthesis of π-conjugated phosphonates derivatives of pyridine, in trans configuration. The pyridine moiety was reacted with MeI, BF3, RuCl3, and the phosphonate group did not interfere with those reactions, which is very important for further uses in material sciences. Work is in progress to graft these compounds at the surface of metal oxides and to study the optical properties of the resultant organic-inorganic hybrids.