

1 Introduction
Inorganic open-framework materials with regular pore architectures are an important class of functional materials because of their widespread applications in catalysis, separation, and host–guest assemblies [1–3]. In the early 1980s, a new class of zeolite-like molecular sieves (denoted AlPOs) was first discovered by Wilson et al. [4]. Since then, a large number of aluminophosphates with rich variety of structures have been synthesized in the presence of organic amines as templates or structure directing agents. Most of these compounds have neutral open-frameworks with Al/P ration of 1, whose structures are constructed from strict alternation of AlO4 and PO4 tetrahedra. A variety of metal or transition metal ions can be incorporated into the frameworks of AlPOs through substituting part of Al sites to form many metal aluminophosphates (MeAPOs) with unusual catalytically activities and Brönsted acid sites [5]. Among the MeAPOs, iron-substituted aluminophosphates (FAPOs) have received considerable interest due to their magnetism characters and efficient chemical activities for some specific catalytic reactions (i.e. cyclohexane oxidation) [6,7].
Al atoms in the structures of AlPOs and MeAPOs are often found in a tetrahedral coordination, and a few can be mixed-bonded adopting four, five, and/or six coordinations with oxygen atoms as in the cases of AlPO4-12 [8], AlPO4-21 [9], AlPO4-17 [10] and AlPO4-CJ2 [11]. That all Al atoms are five-coordinated has been reported in only two compounds K[Ni(H2O)2A12(PO4)3] [12] and NH4[Co(H2O)2A12(PO4)3] [13]. In FAPO structures, the oxidation state of Fe atoms are observed as +3, +2 or mixed valences [6,7,14–18], but these atoms are often in the same coordination environments. Up to now, the metal aluminophosphates contain mixed-valence irons with different coordination states have not been reported.
Here, we describe the crystal structure of a novel iron aluminophosphate [FeII(H2O)2Fe0.8IIIAl1.2(PO4)3]·H3O (I). Its structure is closely related to that of nickel aluminophosphate K[Ni(H2O)2A12(PO4)3] and cobalt aluminophosphate NH4[Co(H2O)2A12(PO4)3]. However, the mixed-valence iron atoms (FeII/FeIII in the ratio of 5/4) and their particular coordination environments in I make it interesting from both structure and property point of view.
2 Experimental section
2.1 Synthesis
The synthesis of I was carried out using solvothermal method in a gel with molar composition 1.0 (i-PrO)3Al: 0.5 FeCl2·4 H2O: 4.1 H3PO4: 4.8 imidazole: 12.0 triethylene glycol (TEG). The typical synthesis procedure was as follows: 2.0 g of (i-PrO)3Al was first dispersed into 16 ml of TEG with stirring, 3.2 g of imidazole and 2.72 ml of phosphoric acid (H3PO4, 85% in water) was then added, the mixture was stirred until it was homogeneous. Finally, 0.97 g of FeCl2·4H2O was added to the above reaction mixture. The reaction mixture was further stirred for 1 h, and then was sealed in a Teflon-lined stainless steel autoclaves and heated in an oven at 180 °C for 5 days under static conditions. Deep purple-red rhombus crystals of I crystallized in a yield of 15% based on iron together with light khaki byproducts (an iron–aluminum phosphate with LAU structure type). A batch of crystals of I were carefully separated under the optical microscope for the further analysis.
2.2 Compositional analyses
Inductively coupled plasma (ICP) analysis was performed on a Perkin–Elmer Optima 3300 DV ICP instrument, which gave the contents of Al as 6.93 wt.%, Fe as 20.95 wt.% and P as 20.57 wt.% (calcd Al: 6.85; Fe: 21.32; P 19.67). Elemental analyses conducted on a Perkin–Elmer 2400 elemental analyzer indicated that no organic molecules or NH4+ ions were occluded in I.
2.3 Thermal analysis
TG-DTA measurements were obtained using a NETZSCH STA 449C unit with a heating rate of 10 °C/min under air atmosphere up to 800 °C. As shown in Fig. 1, the thermogravimetric analysis gave one step weight loss of 12.5 wt.% (calcd 11.6 wt.%) from 350 to 500 °C, which corresponded to the remove of water molecules in I. The further XRD analysis indicated that the structure of I was collapsed and transformed to dense phase after it calcinated at 400 °C for 2 h to remove water. The 57Fe Mössbauer Spectrometry of the calcined sample showed that all of the iron atoms are Fe(III).
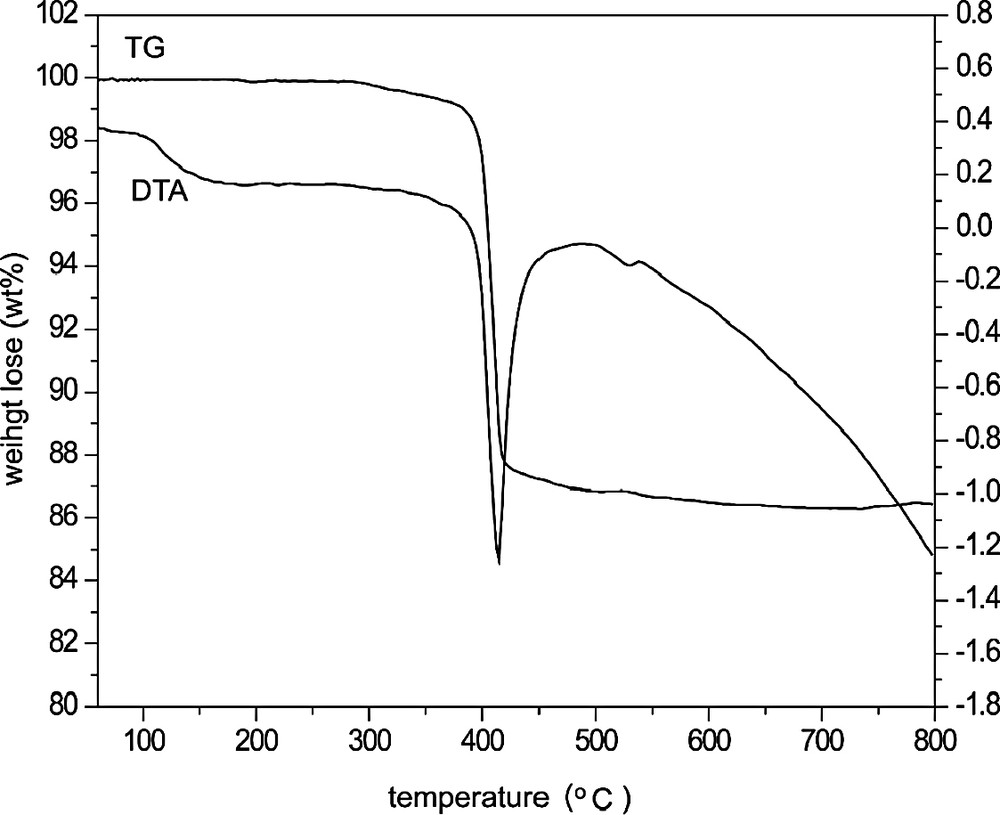
TG-DTA curve of I.
2.4 Single-crystal X-ray diffraction analysis
A suitable single crystal with dimensions of 0.15 × 0.15 × v0.03 mm3 was carefully selected and glued onto a glass fiber for single-crystal X-ray diffraction analysis. The intensive data were collected on a Siemens SMART CCD diffractometer using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) at a temperature of 20 ± 2 °C. Data processing was accomplished with the SAINT processing program [19]. The structure was solved in the space group C2/c by direct methods and refined by a full-matrix least-squares approach on F2 by SHELXTL software package [20]. The partial occupancy of aluminum by iron on the site of five-coordinated atom was not considered at first. A full occupancy of the aluminum on this site led the thermal parameter to zero, while a full occupancy of iron on this site led to the thermal parameters of others atoms converging to zero. Then the occupancy factor of this site was refined and converged to a ratio close to 60:40 for Al/Fe in order to be in agreement with the chemical analysis (Al/Fe ratio of 2/3) of I. Crystal data and details of the data collection are listed in Table 1. Table 2 presents the final atomic coordinates and isotropic thermal parameters. The selected bond lengths and bond angles are listed in Table 3.
Crystal data and structure refinement for compound I
| Empirical formula | H7 Al1.20 Fe1.80 O15 P3 |
| Formula weight | 472.87 |
| Temperature | 293(2) K |
| Wavelength | 0.71073 Å |
| Crystal system, space group | Monoclinic, C2/c |
| Unit cell dimensions | a = 13.3200(14) Å a = 90° |
| b = 10.2104(11) Å β = 108.590(2)° | |
| c = 8.8412(9) Å γ = 90(3) | |
| Volume | 2436.97(11) Å3 |
| Z, Calculated density | 4, 2.756 Mg/m3 |
| Absorption coefficient | 2.906 per mm |
| F(000) | 938 |
| Crystal size (mm) | 0.15 × 0.15 × 0.03 |
| Theta range for data collection | 2.57–28.18° |
| Limiting indices | –17 ≤ h ≤ 13, –13 ≤ k ≤ 12, –11 ≤ l ≤ 11 |
| Reflections collected/unique | 3422/1306 [R(int) = 0.0352] |
| Completeness to theta = 28.18 | 93.4% |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 1306/1/104 |
| Goodness-of-fit on F2 | 1.174 |
| Final R indices [I > 2σ(I)] | R1 = 0.0525, wR2 = 0.1155 |
| R indices (all data) | R1 = 0.0611, wR2 = 0.1186 |
| Largest diff. peak and hole | 0.839 and –0.555 e/Å3 |
Atomic coordinates (× 104) and equivalent isotropic displacement parameters (Å2 × 103) for compound I
| x | y | z | U(eq) | |
| Fe(1)a | 0 | –2124(1) | –2500 | 12(1) |
| Fe(2)b | –1703(1) | –4241(1) | –742(1) | 12(1) |
| Al(1)c | –1703(1) | –4241(1) | –742(1) | 12(1) |
| P(1) | 0 | –4940(2) | –2500 8(1) | |
| P(2) | –2907(1) | –6258(1) | 3297(2) | 10(1) |
| O(1) | –717(3) | –4001(3) | 1955(4) | 12(1) |
| O(2) | –2092(3) | –5817(3) | 1704(4) | 13(1) |
| O(3) | –610(3) | –4166(4) | 1133(4) | 13(1) |
| O(4) | –2327(3) | –2716(3) | –1571(4) | 14(1) |
| O(5) | –2728(3) | –4510(3) | 324(4) | 13(1) |
| O(6) | –971(3) | –1016(4) | –1694(5) | 18(1) |
| O(7) | 1136(4) | –1983(5) | –128(6) | 25(1) |
| O(1W) | 0 | 1376(8) | –2500 | 57(2) |
a U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
b Occupancy factor fixed at 40%.
c Occupancy factor fixed at 60%.
Selected bond lengths (Å) and angles (°) for compound I
| Fe(1)–O(6)#1 | 2.012(4) |
| Fe(1)–O(6) | 2.012(4) |
| Fe(1)–O(7)# | 12.165(5) |
| Fe(1)–O(7) | 2.165(5) |
| Fe(1)–O(1) | 2.260(4) |
| Fe(1)–O(1)# | 12.260(4) |
| M–O(4) | 1.806(4) |
| M–O(2) | 1.817(4) |
| M–O(3) | 1.825(4) |
| M–O(5) | 1.910(4) |
| M–O(1) | 1.957(3) |
| P(1)–O(3)#2 | 1.526(4) |
| P(1)–O(3)#3 | 1.526(4) |
| P(1)–O(1) | 1.535(4) |
| P(1)–O(1)#1 | 1.535(4) |
| P(2)–O(6)#4 | 1.513(4) |
| P(2)–O(5)#2 | 1.531(4) |
| P(2)–O(4)#4 | 1.533(4) |
| P(2)–O(2) | 1.546(4) |
| O(7)–H(7A)...O(2)#3 | 2.824(6) |
| O(7)–H(7B)...O(5)#7 | 2.905(6) |
| O(6)#1–Fe(1)–O(6) | 111.6(2) |
| O(6)#1–Fe(1)–O(7)#1 | 87.09(17) |
| O(6)–Fe(1)–O(7)#1 | 88.62(17) |
| O(6)#1–Fe(1)–O(7) | 88.62(17) |
| O(6)–Fe(1)–O(7) | 87.09(17) |
| O(7)#1–Fe(1)–O(7) | 172.4(3) |
| O(6)#1–Fe(1)–O(1) | 156.23(14) |
| O(6)–Fe(1)–O(1) | 92.18(14) |
| O(7)#1–Fe(1)–O(1) | 92.90(16) |
| O(7)–Fe(1)–O(1) | 93.57(15) |
| O(6)#1–Fe(1)–O(1)#1 | 92.18(14) |
| O(6)–Fe(1)–O(1)#1 | 156.23(14) |
| O(7)#1–Fe(1)–O(1)#1 | 93.57(15) |
| O(7)–Fe(1)–O(1)#1 | 92.90(16) |
| O(1)–Fe(1)–O(1)#1 | 64.08(17) |
| O(4)–M–O(2) | 123.21(17) |
| O(4)–M–O(3) | 117.57(17) |
| O(2)–M–O(3) | 119.21(17) |
| O(4)–M–O(5) | 90.90(16) |
| O(2)–M–O(5) | 87.75(16) |
| O(3)–M–O(5) | 92.67(16) |
| O(4)–M–O(1) | 88.40(15) |
| O(2)–M–O(1) | 89.74(16) |
| O(3)–M–O(1) | 90.71(16) |
| O(5)–M–O(1) | 176.48(16) |
| O(3)#2–P(1)–O(3)#3 | 106.5(3) |
| O(3)#2–P(1)–O(1) | 113.24(18) |
| O(3)#3–P(1)–O(1) | 110.64(19) |
| O(3)#2–P(1)–O(1)#1 | 110.64(19) |
| O(3)#3–P(1)–O(1)#1 | 113.24(18) |
| O(1)–P(1)–O(1)#1 | 102.7(3) |
| O(6)#4–P(2)–O(5)#2 | 108.5(2) |
| O(6)#4–P(2)–O(4)#4 | 112.5(2) |
| O(5)#2–P(2)–O(4)#4 | 110.5(2) |
| O(6)#4–P(2)–O(2) | 111.3(2) |
| O(5)#2–P(2)–O(2) | 109.5(2) |
| O(4)#4–P(2)–O(2) | 104.6(2) |
| P(1)–O(1)–M | 133.4(2) |
| P(1)–O(1)–Fe(1) | 96.59(17) |
| M–O(1)–Fe(1) | 128.73(17) |
| P(2)–O(2)–M | 134.2(2) |
| P(1)#3–O(3)–M | 139.7(2) |
| P(2)#5–O(4)–M | 142.1(2) |
| P(2)#6–O(5)–M | 129.5(2) |
| P(2)#5–O(6)–Fe(1) | 130.7(2) |
2.5 57Fe Mössbauer spectrometry
The 57Fe powder absorption Mössbauer spectra were recorded by a conventional constant acceleration Mössbauer spectrometer with a 57Co/Pd source at room temperature. All isomer shift values (d) given hereafter were referred to as α-iron. Experimental data were resolved into symmetric quadrupole doublets with Lorentzian lineshapes using an iterative least-square fit program.
3 Results and discussion
3.1 Structure description
Each asymmetric unit of I, as shown in Fig. 2, contains 12 crystallographically independent non-hydrogen atoms including 1 Fe, 1 M with Al/Fe ratio of 3/2, 2 P and 8 O atoms. Fe(1) is six-coordinated on the twofold axis, which is linked to two equivalent PO4 tetrahedra via two bridging O(6) atoms (Fe(1)–O(6) = 2.012(4) Å) and two H2O molecules (Fe(1)–OH2 = 2.165(5) Å) in a trans-position. The remaining two vertices are two μ3-O atoms (Fe(1)–O(1) = 2.260(4) Å) that are bonded to two AlO5 trigonal bipyramids and a PO4 tetrahedra sharing one edge with Fe(1). The O–Fe–O bond angles are in the range of 64.08–172.4°, indicating the FeO4(H2O)2 octahedron is highly distorted. Bond-valence calculation [21] results suggest that the Fe(1) is present as Fe2+. M is five-coordinated to four bridging oxygen and one μ3-O atom, making five M–O–P bonds and one M–O–Fe bond. The M–O bond length (1.806(4)–1.957(3) Å) is clearly longer than the Al–O distances (1.770–1.927 Å) in the related structures of K[Ni(H2O)2A12(PO4)3] [12] and NH4[Co(H2O)2Al2(PO4)3] [13]. This is caused by 40% of Al substituted by Fe on this position based on the ICP analysis. Both two distinct P atoms with of P(1) on the twofold axis are tetrahedrally coordinated by oxygen atoms connecting to M and Fe atoms. The P–O bond lengths (1.513(4)–1.546(4) Å) and O–P–O bond angles (102.7(3)–113.24(18)°) are within the expected ranges.

Thermal ellipsoid plot (50% probability) and atomic labeling scheme of I.
The linkages of FeO4(H2O)2 octahedra, MO5 trigonal bipyramids and PO4 tetrahedra through oxygen form the open-framework of I, which contains 8-membered ring (MR) channels along the [001] direction (Fig. 3). It consists of anionic framework of formula [FeII(H2O)2FeIII0.8Al1.2(PO4)3]–, whose negative charges are compensated by the protonated H3O+. Considering the charge neutrality of I, the five-coordinated Fe should has an oxidation state of +3. This is further confirmed by the Mössbauer study as discussed below. The anionic framework of I is similar to those of nickel aluminophosphate K[Ni(H2O)2Al2(PO4)3] and cobalt aluminophosphate NH4[Co(H2O)2Al2(PO4)3]. Its framework can be described as a series of 2D 4,8-net sheets (Fig. 4) formed by alternating MO5 trigonal bipyramids and PO4 tetrahedra, which are further connected together by P(1) and FeII(1) coordinated two H2O molecules. One protonated H3O+ molecule locates in each 8-MR channel (Fig. 3) and forms 2 weak H-bonds with the oxygen atoms in the framework. The H-bonding distances are 2.824(6) Å for O(7)–H(7A)···O(2) and 2.905(6)Å for O(7)–H(7B)···O(5).
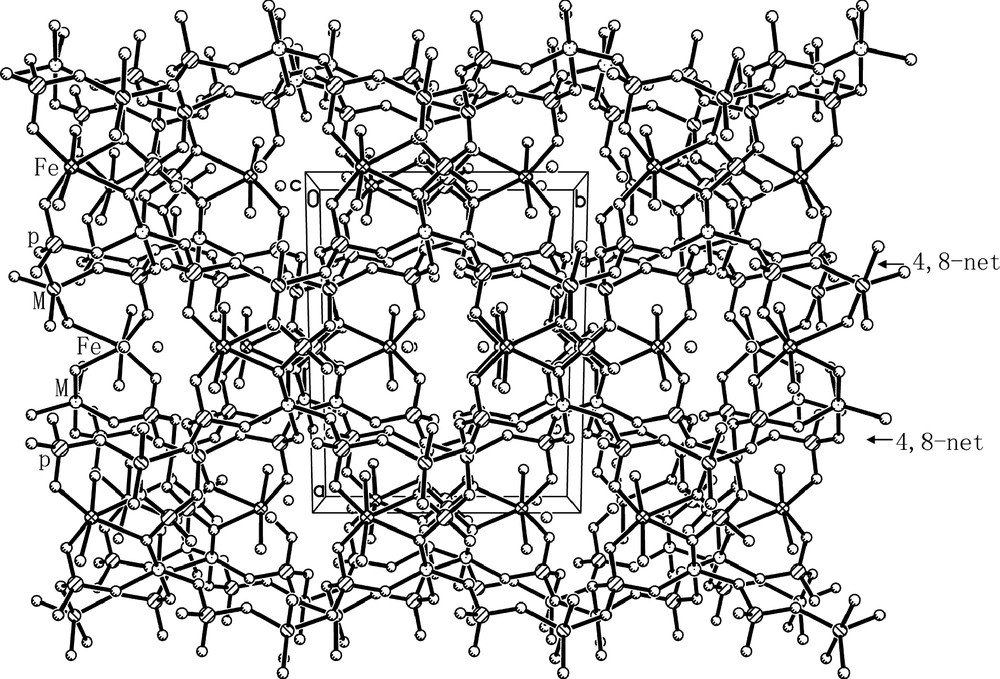
Projected view of the framework of I along the [001] direction shows the 8-MR channels and water locations.
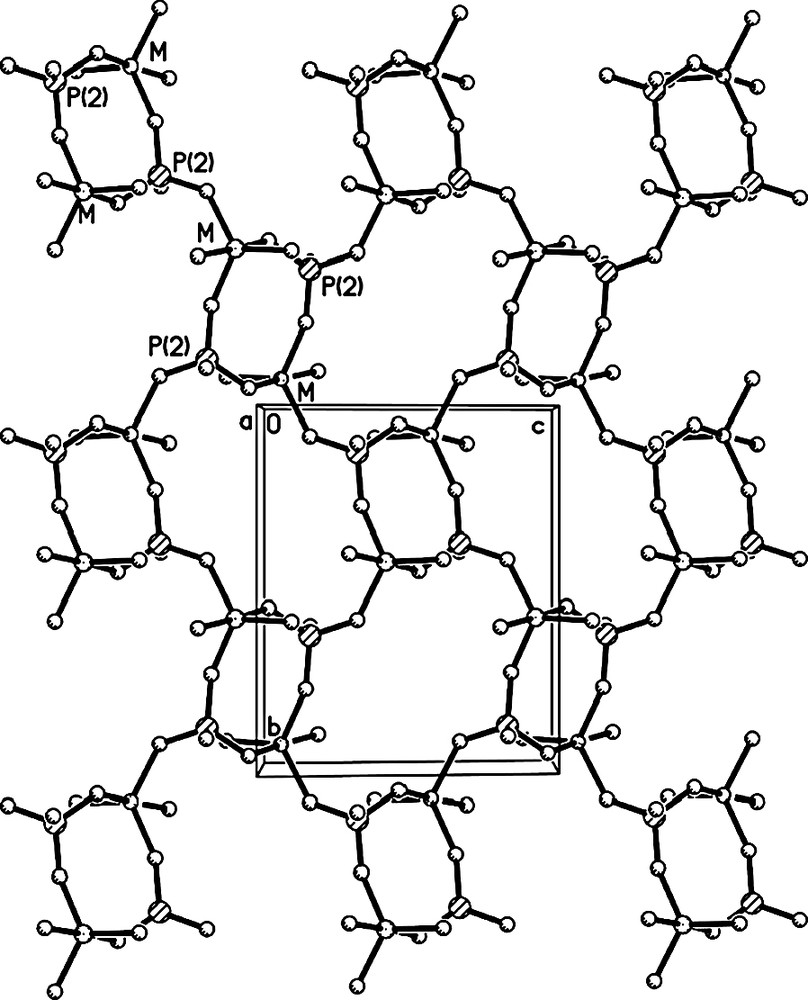
The 4,8-net sheet.
3.2 57Fe Mössbauer spectrometry
57Fe Mössbauer spectroscopy can help to determine whether iron atoms are Fe(II), Fe(III) [22–25] or mixed-valence [26–35]. The room-temperature Mössbauer powder spectrum of the sample (Fig. 5) indicates the presence of pure electric quadrupole interactions, and the spectrum was fitted by means of classical symmetric Lorentzian doublets. The fit obtained from a decomposition of the spectrum can be divided into two doublets (as seen in Table 4). The isomer shift and quadrupole splitting for narrow one are 0.33 and 0.34 mm/s, respectively, which are typical for high-spin trivalent iron. While the value for the wide one is corresponded to high-spin divalent iron [36]. Furthermore, the observed relative intensities of two doublets lead to Fe3+/Fe2+ ratio of 44.65/55.35, closed to 4/5.

Mössbauer spectra of I at room temperature.
Mössbauer parameters for compound I
| Subspectrum | IS (mm/s) | QS (mm/s) | W (mm/s) | Rel. area (%) |
| Narrow doublet | 0.33 | 0.34 | 0.21 | 44.65 |
| Wide doublet | 1.24 | 2.89 | 0.17 | 55.35 |
Up to now, three transitional metal elements Fe, Co and Ni have been successfully incorporated into this kind of octahedron–trigonal bipyramid–tetrahedra framework. The iron substituted case is particularly interesting because it contains the mixed-valence Fe(II) and Fe(III) atoms. The formation of Fe(II)–O–Fe(III) bond and the unusual coordination environments of iron atoms will cause the interesting magnetic and electronic properties of I. Further work is under going.
5 Acknowledgments
We are grateful to the National Natural Science Foundation of China and the State Basic Research Project (G2000077507) for financial supports.
The supplementary material has been sent to the Fachinformationzentrum Karisruhe, Abt. PROKA, 76344 Eggenstein-Leopoldshafen, Germany, as supplementary material CSD-number 414052 and can be obtained by contacting the FIZ.