

1 Introduction
Coordination chemistry especially of the transition metals is an important area of chemical research as it shows tremendous interdisciplinary relevance, e.g. for catalysis and for bioinorganic chemistry including even aspects of medicine regarding therapy as well as diagnosis [1–4]. In this context, coordination chemistry studies under confined conditions seem to be of special importance. Further on, we have to realize that in biological cells, the ‘water’ in between biomolecules only exists in the form of a small number of H2O layers. Consequently, to understand the structure and behaviour of water assemblies – either pure or mixed with some electrolytes – under confined conditions, i.e. at a nanometre scale, is a fundamental challenge shared by chemists, physicists, and biologists. In this context, there is – generally speaking – a considerable interest to design easily accessible synthetic model systems capable of mimicking some basic related key-role functions in living systems.
The question arises which properties a related artificial cell should have. First, some kind of frontier/barrier (membrane function) should exist that allows a clear differentiation between the outer macrocosm characterized by bulk properties (solvent) and the inner microcosm characterized by the existence of structures/functions, which are limited in size and subjected to quantum-mechanical laws. Second, specific gates that may be opened or closed on demand should be available in order to allow for exchange processes between exterior and interior and their control (‘feeding’ function). Third, opening and closing the gates should be dependent on the formation of non-covalent bonds, e.g. hydrogen bonds insuring a not too strong but not too weak ‘mechanical device’ (‘screening’ function). Fourth, a cavity large enough to get filled with water molecule assemblies should be available for handling encapsulated species via hydrogen bonding, ion pairing, hydrophobic interactions or coordinative links (‘assimilation’ function). Fifth, the artificial cell should preferably be self-assembled in solution in order to generate in only one single step a very large number of identical vessels (very formally a replication function). This would allow to study straightforwardly what happens with respect to cell-environment interactions, e.g. by NMR in solution directly, or by X-ray diffraction studies of the final ‘aggregated’ artificial cells (into a crystalline solid) after the solution events have taken place, though the latter method is not the optimal choice as crystallization processes can influence the results.
It was now possible to construct artificial cells fulfilling the above-mentioned categories. They are characterized by sizeable water-filled cavities, tunable pores/channels, flexible tunable interiors [5–7], and will be discussed here. They are based on a rather robust skeleton (Pentagon)12(Linker)30 ≡ [{(Mo)Mo5O21(H2O)6}12{Mo2O4(ligand)}30]n– [8], which can be modified in various routes and may easily be synthesized even on a (kilo)gram scale [9].
Quite recently, it was demonstrated using multinuclear NMR techniques and Raman spectroscopy that such nanocapsules are quite stable both in aqueous and non-aqueous solvents [10,11], and that they are characterized by a chameleonic behaviour of water molecules trapped under different conditions, quite reminiscent of the low-density/high-density duality typical of biological systems [12]. Since the ability of special polyoxometallate systems to mimic some special aspects of biological chemistry-related processes is well recognized now [12,13], we propose here an illustrative and therefore of course simplified concept that refers to the option of studying different types of interactions of cations with molybdenum-oxide-based artificial cells, in particular with encapsulated water. A special aspect refers in this context to the well-known Hofmeister series effect, and the related behaviour of water on interfaces [14].
2 Artificial cell or nano-chromatograph (AC/NC)?
The perforated ‘membranes’ {P12L30} of the considered AC/NCs (Fig. 1) are formed by self-assembly in solution from pentagonal units (P) exhibiting a central pentagonal O = MoVIO5(H2O) bipyramid sharing five equatorial edges with O = MoVIO4(H2O) octahedra, and formally linkers (L) {O2(MoV=O)(μ-O)2(MoV=O)O2X} involving dimeric edge-sharing octahedra [5,8]. In the linker unit, X stands for bidentate HCOO– (FORM), CH3COO– (ACET), H2PO2– (HYPO), SO42– (SUL) or PO43– (PHOS) ligands/ions, which can be used to tune the hydrophilic/hydrophobic behaviour as well as the coordinating strength of the interior walls.
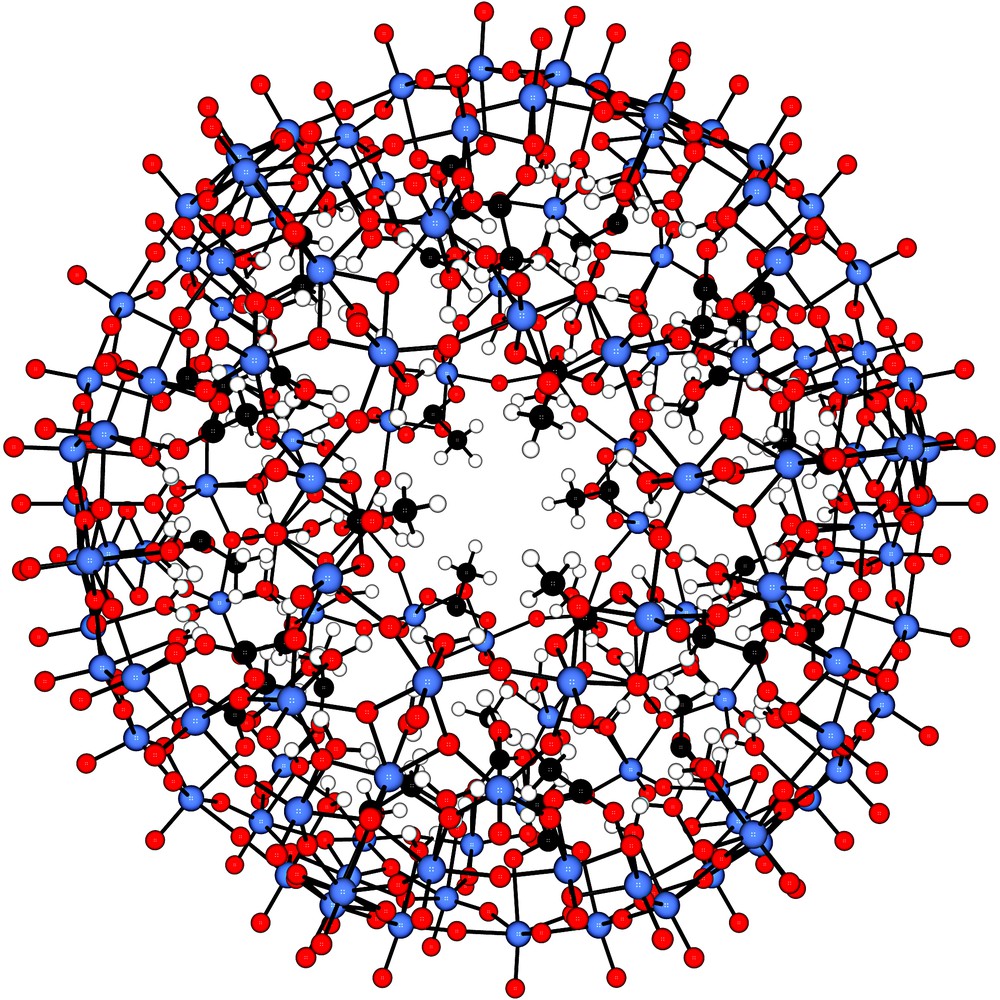
View down the C3-axis evidencing the pore structure of the spherical artificial cell/nano-chromatograph [P12{Mo2VO4ACET}30]42– with the pentagonal-shaped unit P = {(Mo)Mo5O21(H2O)6}6– (definition used throughout the paper) (ACET = CH3COO–). Other nanocapsules may be easily derived from this basic species being stable in solution, by substituting under acid conditions the acetate ligands by other anions such as formate, hypophosphite, phosphate, or sulphate. The water assembly trapped inside the cavity is not shown for better clarity, but its structure was found to be strongly dependent on the kind of anionic bridging ligand as constituent coordinated to the edge-sharing di-octahedral linker units. Colour code: red = oxygen, blue = molybdenum, black = carbon and white = hydrogen (for references, see text).
Let us anticipate what could happen to cations (CAT) ‘travelling’ – in the context of an illustrative view – towards the negatively charged nanocapsules: first they will encounter the typical spherical outer structure of the inorganic hydrophilic membrane responsible for the very high solubility of the nanocapsules in water, which is caused by the hydrogen bonding between the spikes (mainly outer Mo=O, but also Mo–OH2 units) and surrounding water molecules of the (bulk) solvent. A first possible subsequent event for the cations could be to interact with these spikes, forming either an outer-sphere association CAT–OH2···O=Mo or an inner-sphere CAT–O=Mo complex. As we know that these Mo=O bonds display comparably low electronic densities, i.e. less basic sites (in agreement with the PACHA model [15]), one may tentatively assume that such associations would be easily thermally broken corresponding to the high surface mobility for the ‘adsorbed’ cations. This kind of behaviour may be called Molecular Wandering at the Outer Interface (MWOI) in order to distinguish it from the other possibility Molecular Sticking at the Outer Interface (MSOI), thus describing a situation where cations are unable to move easily due to being stuck at the spikes.
During ‘exploration’ of the outer side of the inorganic membrane via the (thermally activated) Brownian motion, translocation of the MWOI cations towards the pores may be observed. A following anticipated event could be rejection of the cations outside the pores (with the consequence of either going directly into solution or back to the outer Mo=O shell groups), if they are too big to fit into the pores or channels for ‘successful communication’ with the inner part of the nanocapsules. Another possibility would be a more or less good complementarity between the size/shape of the cations and that of the pores, allowing fixation/complexation by the oxygen atoms of the 20 pores, a situation which is remarkably similar to that of the crown ethers. This kind of event should be called as usual in supramolecular chemistry ‘Molecular (Pore) Recognition’ (MPR) and would lead ultimately to the formation of closed nanocapsules. Yet another possibility may be visualized with hydrated cations displaying a quite labile hydration shell. After transitory removal of their hydration shell, many cations are small enough to enter the channels interacting with the inner part of the AC/NCs. At this stage, different kinds of molecular events may be anticipated. First, it is possible that a complementarity exists between the sizes of the (naked) cations and the channel allowing favourable contacts, and/or that the functionalities of the channel interior are appropriate to trap the cations. This kind of event may be called Molecular Channel Recognition (MCR). (Note that the cation could be considered as only nearly naked, as one or two water molecules could be left in the coordination sphere.) When cations are too small to develop favourable attractive contacts, they are expected either to go backwards (a typical MWOI behaviour) or definitively to cross the channel in order to recompose partially their lost hydration shell with oxygen donors that are available within the cell-interior functions of the AC/NCs and within the water molecule aggregates present inside the central cavity. For such encapsulated cations, a first possibility would be to ‘explore’ – speaking again in an illustrative picture – through Brownian motion the inner walls of the cavity searching for nucleophilic sites displaying enough electronic density. If such sites are available (typically if sulphate or phosphate ligands are present), complexation may occur, leading to inner-sphere ion pairing/complexation, a situation that may be nicknamed Molecular Complexation at the Inner Interface (MCII). In this situation, the coordination shell of the cations is heterogeneous, with one part coming from the ingredients of the internal capsule shell and the other part coming from the water molecule assemblies/shells trapped within the cell cavity. When oxygen donor sites are not available (typically if formate, acetate or hypophosphite bridges are present), cations are expected to develop interactions preferentially with encapsulated water assemblies. Here a first possibility could be encapsulation of guest cations in clathrate-type host cages, an event that may be called Molecular Encapsulation through Clathrate Formation (MECF). A second possibility could be a ‘type of copolymerisation’ of the hydrated guest cations and the encapsulated water molecules through hydrogen bonding, leading to unprecedented mixed water electrolyte assemblies, an event that may be called Molecular Recognition of Water Structure (MRWS). A last possibility would be a complete breakdown of the water structure leaving more or less amorphous/chaotic water assemblies, an event nicknamed Molecular Breakdown of Water Structure (MBWS).
Obviously, this identification of eight different types of behaviour is somewhat arbitrary and idealized, in agreement with the simplified illustrative view mentioned in the title. One may easily visualize several intermediate situations corresponding to partial dehydration of the cations or thermally activated molecular hopping. All these processes of course strongly depend on the temperature of the solution.
In the present case, we may refer to four fundamental parameters that should govern cationic behaviour towards the AC/NCs:
- • The first crucial parameter that refers to a rejection threshold for solute species is the size of the pores. For the AC/NCs, the radii of the channels were estimated using a standard set of van der Waals radii [16] and available crystal structure data. The average following channel radii were found: 1.51 Å for ACET [17a], 2.30 Å for FORM [17b,c], 1.45 Å for HYPO [18] (assuming a P–H bond length of 1.2–1.25 Å), 1.31 Å for SUL [7] and 1.22 Å for PHOS [6]. Obviously, these values give an idea of the free space available at the narrowest zone of the channels defined by the relative positions of the three (bridging) ligands of the three rather stable {Mo2VO4X} units/linkers (see above and below). It is worth noting that in case of FORM, PHOS, HYPO and SUL ligands, the corresponding channel radii are rather well defined, at least in cases where no partial pore/channel opening according to a related kind of flexibility is given (note: the bidentate ligands can, in principle, ‘swing’ perpendicular to the Mo–Mo bonds, especially at higher temperatures).
A different complex case is the ‘real channel radius’ of the ACET nanocapsule, which is highly dependent on the relative orientations adopted by the three freely rotating methyl groups (assumed C–H bond length of 1.10 Å). In this case, the radius should be at least 0.84 Å if all H-atoms point towards the channel, but not larger than 2.36 Å if no H-atoms point towards the channel. The obvious limitation of this procedure refers to the fact that during host-guest (cation) interactions, the pores might, in principle, be partially opened.
- • The second important parameter is the cation size (referring to inorganic as well as small organic [19]). As the computed channel radii are of the order of magnitude of the radius of a water molecule (1.42 Å for pure water and 1.39 Å for water near an ionic species [20]), cationic species should be mostly dehydrated in order to be able to cross the channels (two water molecules both pointing in direction of the channel might be left). In contrast, neutral water molecules are allowed to move more or less free between the interior and the exterior of the nanocapsules so that we can refer to the presence of water channels. In the following, we have used a set of published effective close-packing ion volumes vJ [21] allowing to compute realistic naked ionic radii ri = (3 vJ/4 π)1/3.
- • As a nearly full dehydration of the cations is required to move across the channels, another important parameter is the lability of the water molecules in the hydration shell. As illustrated in Fig. 2, this lability is highly cation-dependent, with exchange frequencies ranging from even more than 10 billion per second (alkaline cations) down to circa one water molecule exchange per month (Cr3+ ion) [22,23].
- • In addition, it should be mentioned that the overall formal charge of the capsules has of course an influence on the uptake process. For instance, it was observed that the presence of small divalent main group cations like Ca2+ does not necessarily lead to an uptake, as the hydrated cations are found above the pores in case of a capsule with linkers containing low negatively-charged ligands, in contrast to the situation with sulphate ligands where the cations are taken up (valid only under similar channel conditions).
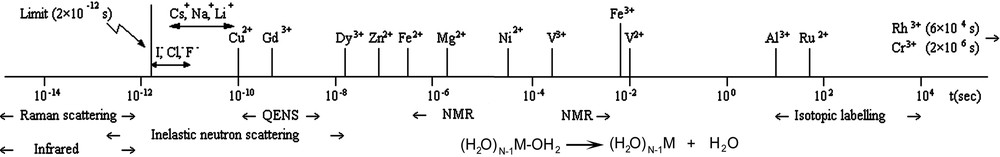
Lability of water molecules in the coordination sphere of metallic cations Mz+(z ≤ 3) and techniques used for determining related data (note the large order-of-magnitude difference between the most inert cation and the most labile one; taken from ref. [23]).
3 Results
Let us first consider cationic species that are too big and/or too inert to enter the AC/NCs. For instance, a typical MPR behaviour was observed in the case of the guanidinium (gua = [C(NH2)3]+) containing species gua20 ⊂ P12{Mo2VO4SUL}10{Mo2VO4HYPO}20]32– [19] (Fig. 3, top). As the ionic radius of this cation is not available in the literature, it was estimated from the computation of its molecular volume (v ≈ 55 Å3) using standard Monte-Carlo techniques [24] and van der Waals radii [16]. The radius obtained from this estimated volume is 2.36 Å, thus much larger than that of the sulphate- or hypophosphite-based channels (1.31 and 1.45 Å, respectively). However, it agrees perfectly with the openings on the level of the μ-oxo groups of a {Mo9O9} ring/pore (2.36 Å), a fact that explains the MPR behaviour. The organic cation is encapsulated/integrated completely analogue to the known situation of a crown-ether species published by Lehn et al. (for details, see [19]).
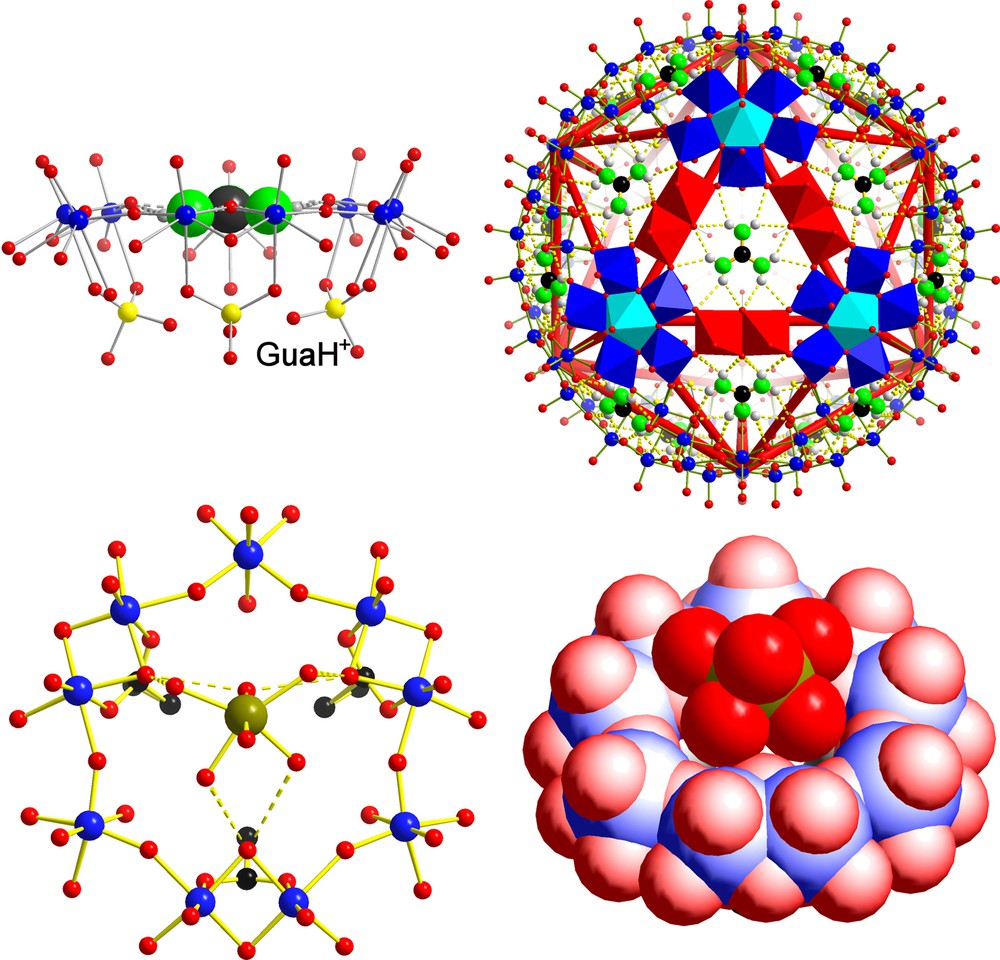
Molecular Pore Recognition (MPR) behaviour in the case of the guanidinium cation, which fits exactly into the pores of the mixed ligand AC/NC according to [gua20 ⊂ P12{Mo2VO4SUL}10{Mo2VO4HYPO}20]32– (top, showing two different presentations; the right part highlights the construction of one pore from three pentagonal units and three linkers in polyhedral representation). This situation is comparable to the case of the known corresponding crown ether compound of J.-M. Lehn et al. (see [19]). In the case of the [P12{Mo2VO4ACET}30]42– AC/NC, the hexaaquo nickel(II) complex sits above the pores (bottom, showing one segment in different presentations). Colour code and definition for P as in Fig. 1, but with sulphur/phosphorus atoms displayed in yellow and nickel atoms in green (for references, see text).
A different kind of Molecular Pore Recognition behaviour of the hydration shell (MPR) has been identified in the case of an acetate-type capsule (2.36 Å maximum pore opening) interacting with [Ni(H2O)6]2+ complexes [6] (Fig. 3, bottom). With a naked ionic radius of 0.78 Å [21], it is evident that the relatively low lability of the Ni2+-ion hydration shell is responsible for the trapping of the whole complex above the {Mo9O9} pores (not within(!) as in case of the gua situation). As the lifetime of a water molecule within the hydration shell of Ni2+ ions is known (32 μs at T = 25°C) [25], we deduce that only cations displaying the water lifetime within their hydration shell (approximately less than 1 μs) could enter the AC/NCs (note: not only in the case of low charged (42–) capsules, but also in case of higher charged capsules (72–), [Ni(H2O)6]2+ was found above the pores [26]). Interestingly enough, owing to its very similar ionic radius and hydration-shell lability, a MPR behaviour was anticipated for magnesium ions, which was indeed found [26]. Consequently, if we are interested in real cation uptake events, the focus of attention has to be directed towards the highly labile cations which are located left to the Ni2+ ions in Fig. 2.
Among the cations displaying larger water-exchange rates, Cs+ is surely a most typical candidate to study, while it could be too large even in a fully dehydrated state (ri = 1.65 Å [21]) to cross the channels of the AC/NC into the interior. This prediction was confirmed in case of a [Cs10 ⊂ P12{Mo2VO4SUL}30]62– AC/NC [6] with a channel radius of 1.31 Å (Fig. 4, top). Here we have a nice example of a MCR behaviour, in contrast to the smaller rubidium cation (ri = 1.49 Å [21]) that was found to display a typical MWOI behaviour and is located at several positions above the pores (Fig. 4; for details, see [6]). In such a case, we obviously have to take into consideration that the final observed position can also be influenced by the events during the crystallization process, especially if the cations do not enter the capsule areas. On the other hand, the sodium ions with ri = 0.98 Å are small and the water of hydration is labile enough to permit them crossing the channel to land in the interior of the AC/NCs. Accordingly, a MCII behaviour (Fig. 5, top) was observed for sodium ions with ‘coordination’ to three oxygen atoms of three sulphate groups in the case of [{(NH3CONH2)20 + Na13} ⊂ P12{Mo2VO4SUL}30]39– AC/NCs [6] as well as in the case of the mixed [Na6 ⊂ P12{Mo2VO4SUL}24{Mo2VO4ACET}6]60– AC/NC [7]. With an ionic radius of 1.33 Å [21], potassium ions provide (as do sodium cations) the interesting case of an almost perfect ‘match’ with the below-channel area, which could be proven in case of [(formamidinium)20 + K5 ⊂ P12{Mo2VO4SUL}30]47– AC/NCs [26]. Experimentally, K+ has been found at a comparable but still ‘lower’ position, as sodium ions at the C3-axes, which was of course expected. Owing to a similar ionic size as sodium (1.06 Å) and the water complex lability (about 1 ns at room temperature [22]), a similar behaviour was expected for the calcium ions as evidenced by the isolation of a [Ca20 ⊂ P12{Mo2VO4SUL}30]32– AC/NC [26] (Fig. 5, bottom). It is worth noting that a typical feature of this kind of MCII behaviour is the hybrid-type six-fold coordination, which was observed for the last two mentioned cations comprising in the coordination sphere three O-atoms from the three sulphate bridges below the {Mo9O9} pore and three O-atoms belonging to the encapsulated water shell/assembly. Considering the cadmium ions, with ionic sizes (1.03 Å) intermediate between those of sodium and calcium ions and characterized by a lower water exchange rate (about 10 ns at room temperature) [22], we have to expect the MCII behaviour which was observed for [Cd4 ⊂ P12{Mo2VO4SUL}30]64–, but interestingly with two different kinds of coordination sites (Fig. 6). The first one is very similar to that mentioned above, i.e. at the C3-axes, but now, four water molecules instead of three enter the coordination polyhedron of the cadmium ions (Fig. 6 left), leading to seven-fold coordination. At the second coordination site (Fig. 6 right), an eight-fold coordination was observed, showing (only) one sulphate group acting as a bidentate ligand as well as eight water ligands belonging to the encapsulated water-shell assembly.
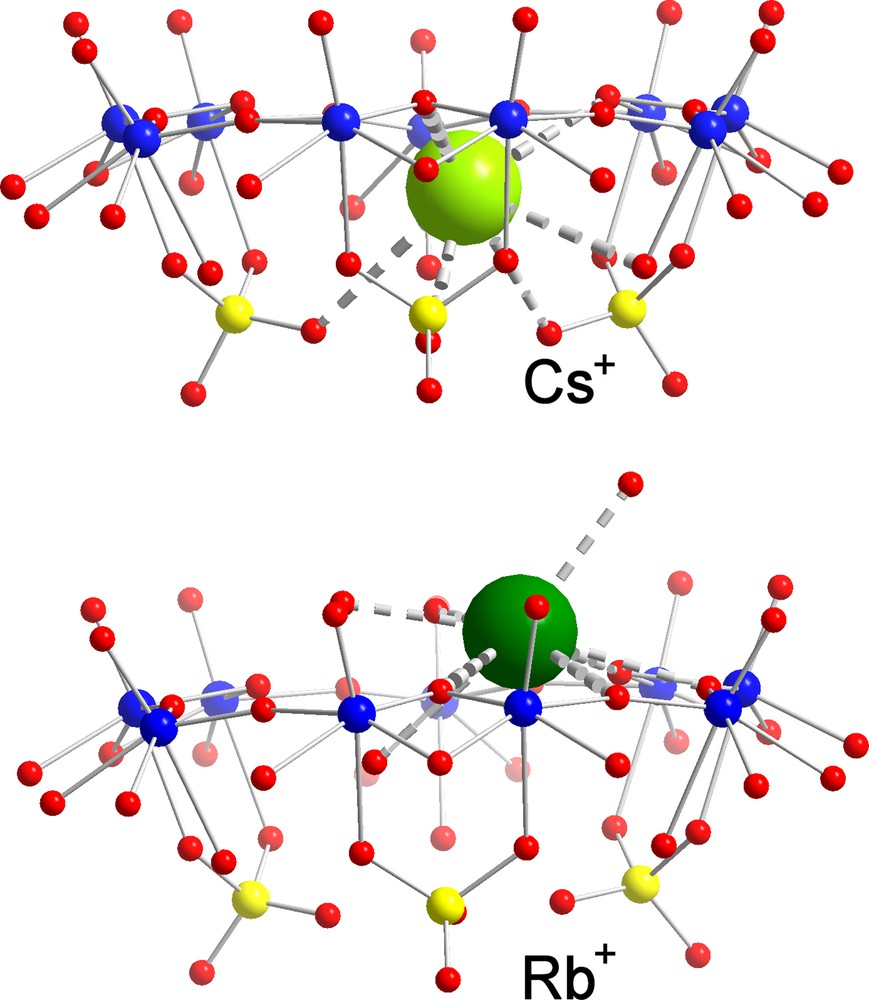
Identification of a Molecular Channel Recognition (MCR) behaviour in the case of [Cs10 ⊂ P12{Mo2VO4SUL}30]62– AC/NC (top) and of a Molecular Wandering at the Outer Interface (MWOI) behaviour of one of the rubidium cations in the case of the same cell (bottom; the corresponding formula cannot be given because of very different Rb+ positions on the cell surface with no full occupation). Colour code as in Figs. 1 and 3, but with caesium and rubidium displayed in green (for references, see text).
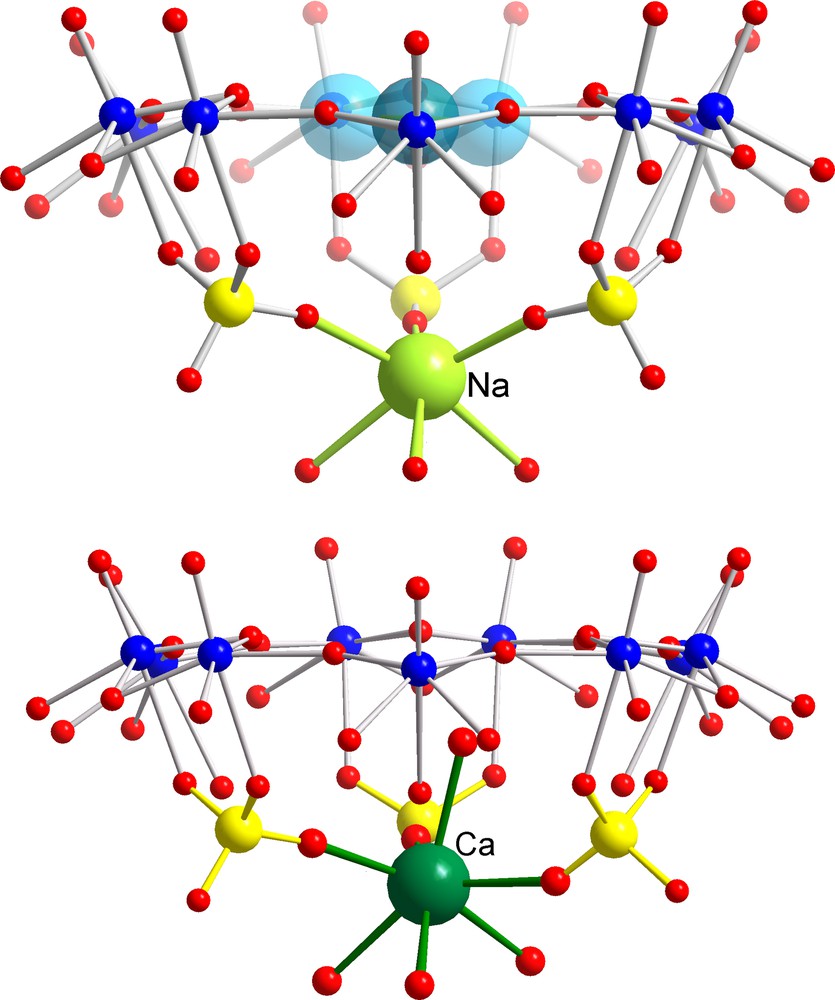
Identification of a Molecular Complexation at the Inner Interface (MCII) behaviour in the case of [{(NH3CONH2)20 + Na13} ⊂ P12{Mo2VO4SUL}30]39– (top; for references, see text) and [Ca20 ⊂ P12{Mo2VO4SUL}30]32– AC/NCs (bottom). The blue spheres correspond to disordered UreaH+ cations in the pores while positioned correspondingly to guanidinium cations, as expected. The potassium situation is similar to the sodium case but of course with longer K–O distances [26]. Colour code as in Figs 1 and 3.

Identification of two different kinds of Molecular Complexation at the Inner Interface (MCII) behaviour in the case of [Cd4 ⊂ P12{Mo2VO4SUL}30]64– AC/NCs [26]. Colour code as in Figs 1 and 3.
This fascinating encapsulation game with the [P12{Mo2VO4SUL}30]72– AC/NCs may be continued one step further by considering trivalent cationic species displaying the same water ‘residence time’ (about 10 ns) as cadmium ions. Referring to Fig. 2, we see that early lanthanide cations, such as Ce3+ (three encapsulated ions) [6] or Pr3+ (four encapsulated ions) [27], are perfectly positioned inside the capsule as their ionic radii (1.18 and 1.16 Å, respectively) are definitely smaller than the typical average channel radius size (1.31 Å). Fig. 7 shows, as expected, that Pr3+ ions are captured within the inner cavity and display exactly the same MCII behaviour as the cadmium ions.
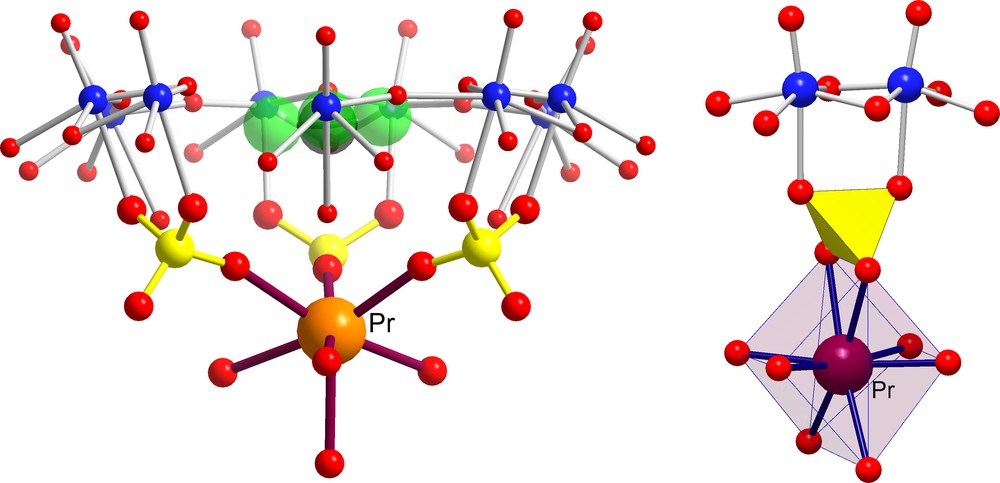
Identification of two different kinds of Molecular Complexation at the Inner Interface (MCII) behaviour in the case of [{(NH2CHNH2)20 + Pr4} ⊂ P12{Mo2VO4SUL}30]40– AC/NCs (for references, see text). The green spheres correspond to formamidinium cations in the pores while positioned correspondingly to guanidinium cations; the few Pr3+ are disordered over several positions. In highly concentrated medium, the cation uptake can be drastically increased [26]. Colour code as in Figs 1 and 3.
4 Special situation with NH4+ ions
Finally, the complicated case of the ammonium ion with an ionic radius of 1.71 Å according to its molecular volume [21] has to be discussed. The special situation refers in particular to the fact that NH4+ can replace H2O in the water polyhedra and can also easily be encapsulated in the centre of the {H2O}20 dodecahedron according to well-known facts [7]. (Therefore it is problematical/impossible to locate these from single crystal X-ray diffraction studies.) Referring e.g. to [P12{MoV2O4HYPO}30]42– AC/NCs, the channel size is too small to permit entrance into the cavity, and correspondingly, the formation of a pure water fullerene-type {H2O}80 assembly is observed [7]. Interestingly, in the case of a [P12{Mo2VO4SUL}30]72– AC/NC, some ammonium cations are in fact found encapsulated within the water assemblies (MECF and/or MRWS behaviour) [7]. Knowing that the channel size (1.31 Å) does not allow entrance after the nanocapsule self-assembly, it probably follows that the ammonium ions were directly encapsulated together with water molecules before the closure of the inorganic shell. (Note that this situation cannot, in principle, be excluded in other cases either, though most of the present results do not support that.)
Here, a crucial point is still the ‘real size’ of the ammonium ion, which is caused by its non-spherical shape and flexible hydrogen bonding options. Based on the fact that a considerable number of ammonium oxy-salts exist that are isostructural with the corresponding potassium salts [28], it is usually assumed that potassium and ammonium ions have approximately the same size. This is typically mentioned in nearly all modern Inorganic Chemistry textbooks. But, according to Shannon, four-fold coordinated potassium ions have a (crystal) ionic radius of 1.51 Å that may ‘increase’ up to 1.78 Å when this ion is surrounded by twelve O- or F-atoms [29]. However, a much smaller value (1.33 Å) is quoted by Pauling in his most famous textbook [30] or computed from ionic volumes according to [21]. Furthermore, based on electron density maps data, intermediate values of 1.44 Å [31] or 1.49 Å [32] have been quoted. (In contrast, on another occasion, Pauling quotes for the ammonium ion a rather low value (1.48 Å) [30], which is more in line with the thermochemical radius (1.43 Å) computed from the empirical Kapustinskii equation [33].) Analysing the N–O distances in a large number of ammonium salts has led to (crystal) radii spanning the 1.63–1.83 Å range [28]. This seems to be in good agreement with the value 1.71 Å calculated from ionic volumes [21] or with the value 1.66 Å derived from electron-density maps [30].
Since it is difficult to locate, as mentioned above, ammonium ions inside the capsules, based on single crystal X-ray structure analyses [7], corresponding 15NH4+ NMR investigations are in progress (E.T.K. Haupt, D. Rehder, A. Müller, to be published).
5 Conclusions and perspectives
Some aspects of general interest for classical aqueous coordination chemistry under confined conditions have been demonstrated. In this respect, it should be realized that there is still no clear final proof of the kind of coordination spheres of several aqua complexes in bulk water, mainly where the second shell and its interaction with the first is concerned [34]. A fascinating aspect refers to the kind of interaction between artificial cells and cations present in (bulk) solutions with many options. First, the materialization in solution of the AC/NC concept allows to some extent to perform tests on the validity of the use of various ionic radii values proposed in the literature in the context of the interpretation of cation channel transport processes. This is due to the fact the AC/NCs offer rather well defined molecular holes (at least under the assumption of rigid pores) for incoming solvated cations. When sufficient data have been accumulated based on systematic variation of the linker ligands and of the (counter-)cations in solution, a scale of ionic sizes relevant for the present type of transportation/uptake processes could be defined. In this context it would be optimal of course to include studies of the processes in solution with NMR, which are currently in progress (see [11] for the uptake and release of Li+), since the conclusions from single-crystal X-ray studies have their limitations as they refer only to final products, i.e. after the crystallization process has taken place, where in principle, changes are possible. In this context, the environment of the cations observed above as well as below the pores may be of interest. The (approximate) scale of sizes based on molecular sieving could also be relevant for solid-state scientists, e.g. those interested in (crystal) nucleation in aqueous solutions as well as for biophysical chemists studying ion transport processes.
As the AC/NCs allow studying nicely how confined water molecules may be coordinated to cationic species, the elucidation of related basic problems/mechanisms leading to inner- or outer-sphere complexes under confined (!) conditions might be of importance especially if one wants to acquire an understanding of the real chemical status of simple only weakly coordinating ions like K+ in prokaryotic or eukaryotic living cells. Note in context with the present confined condition studies: not only macromolecules but also small ions in the cytoplasm are not very flexibly moving and only a few layers of water molecule assemblies interact strongly with biomolecules [35].
Last but not least, a general conclusion is that it is possible to study a versatility of cation structure-directing translocations, which opens new perspectives on the interplay between structure and dynamics on the nanometre scale.
Acknowledgements
A.M. thanks the ‘Deutsche Forschungsgemeinschaft’, the ‘Fonds der chemischen Industrie’, the ‘Volkswagen-Stiftung’ and the European Union for financial support.