

1 Introduction
Future developments in molecular electronics are based on the design of new organic materials displaying electronic and/or optical properties [1]. Particularly light-emitting molecular systems exhibiting highly tunable emission properties are of prime importance for applications in emerging luminescence technologies [2,3]. In the field of conjugated oligomers and polymers, tuning of optical properties has been mainly achieved according to synthetic strategies involving changes in length, substitution, and degree of conjugation [4]. Besides, recognition processes at the molecular level provide an alternative means to control excited-state properties [5]. Especially photosensitive π-conjugated oligomers incorporating a binding unit as recognition site can act as tunable fluorogenic multi-component systems whose fluorescence output is highly sensitive to exogenous chemical species, such as protons or metal ions [6,7]. In this case, protonation or metal binding represents a very simple, reversible process that allows the generation of a variety of emission colors from a limited set of compounds. Conversely, changes in fluorescence emission properties can be utilized for applications in chemosensing or optical switching [8,9].
2,2′-Bipyridine (bpy), 1,10-phenanthroline (phen) and the terdentate 2,2′:6′2″-terpyridine (tpy) represent archetypal polypyridine ligands (Fig. 1) that act as effective chelating agents for a variety of metal ions [10,11]. The photophysics and photochemistry of transition and lanthanide metal ion complexes of these ligands have been the subject of intense investigation for applications in solar energy conversion and luminescent labeling [12,13]. Moreover these ligands were shown to offer remarkable opportunities for the construction of supramolecular architectures or functional nanostructured materials [14,15]. To this end, they served as building-blocks for the metal-assisted self-assembly of sophisticated structures such as helicate or catenates, for examples [5]. Fluorescent receptors were also built that were based on the covalent linkage of a fluorogenic group and a ligand unit through a saturated bridge [16].
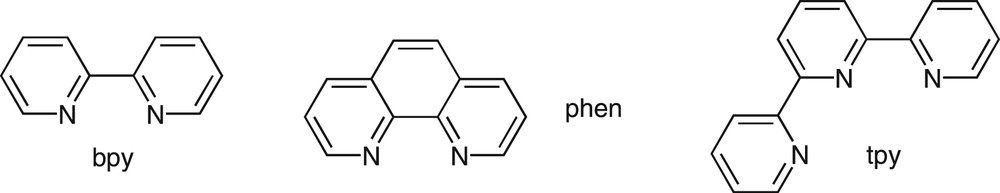
Structures of the parent bpy, phen, and tpy ligands.
The need for optically tunable supramolecular assemblies and materials fostered the research toward efficient modulation of the excited-state properties of polypyridine ligands. It occurred that substituting these heterocycles with π-conjugated molecular building-blocks could represent a strategy of choice toward this purpose. Indeed bpy and phen were shown to be easily introduced at the core of a conjugated backbone without disrupting electronic delocalization, which provides access to highly emissive conjugated oligomers and polymers. Instead, tpy ligands, due to their specific geometrical features, were conveniently introduced at the termini of conjugated molecular rods [17]. This account is intended to give an overview on the photophysical properties of some selected polypyridine ligands containing π-conjugated substituents that appeared recently in the literature. Of the many interesting features exhibited by these systems, the remarkably high tunability of their luminescence properties, such as emitted color or excited-state lifetime, renders them versatile light-emitting units to be incorporated into chemosensors as well as functional materials. Moreover π-conjugated molecules that contain transition metals not only give valuable insight into the interaction of the metal with the delocalized π-electron system, but also allow the design of materials with unique optical, electronic and magnetic properties [18]. As bpy occupies a central place owing to its rich chemistry, most examples reported therein are based on the bpy unit, but some examples of systems based on phen and tpy skeleton are described as well.
2 Conjugated polypyridine ligands with enhanced fluorescence emission
2.1 The case of the parent ligands
The parent bpy is nonfluorescent in nonaqueous solution [19]. This is due to the presence of low-lying excited states of n-π* character that predominantly decay via nonradiative pathways such as efficient intersystem crossing and/or internal conversion. Yet fluorescence emission in bpy has been the subject of controversy as it was found that bpy solutions could emit light. Actually, this behavior was attributed to the formation of hydrate species, or even protonated or complexed bpy due to sample contamination [19]. In acidic solution, the monoprotonated bpy is found to fluoresce whereas the diprotonated species, bpyH22+, is silent due to rapid excited-state deprotonation, which competes with radiative decay to the ground state. Similarly, tpy is nonfluorescent [20]. In contrast, phen is known to exhibit fluorescence emission in solution [21] with a weak quantum efficiency (< 0.01) [22]. Yet the Franck Condon S1 state has been shown to be of π−π* and not n-π* nature [19], on the basis of spectral analogy between the electronic absorption spectrum of phen and that of phenanthrene, the hydrocarbon analog, and also the lack of any solvent polarity effect in the ground state. Actually the allowed π–π* and forbidden n-π* states in phen are so close in energy that they experience strong mixing, which explains not only the low fluorescence quantum yield but also the dependence of fluorescence emission properties on the polarity of the solvent. Similarly to bpy, the monoprotonated phen species is fluorescent and the diprotonated one is not owing to efficient excited-state deprotonation.
2.2 Effect of extended π-conjugation in polypyridine ligands
Clearly these ligands do not represent attractives candidates as intrinsic fluorophores. Recently Joshi et al. [6] showed that the introduction of acetylenic linkages in the three, eight positions of phen provided intensely fluorescent derivatives, 1a–d (Fig. 2, Table 1). The effect was attributed to the increase of conjugation along the three to eight axis, which induces a substantial stabilization of the allowed π−π* state at the expense of the forbidden n-π* state together with an increase of the fluorescence transition dipole moment along this axis. As a result, a series of compounds were synthesized that display high fluorescence quantum yields (Φf = 0.5 – 1) and very short lifetimes (< 3 ns) as typically observed in the case of fully allowed π–π* singlet states (Table 1).
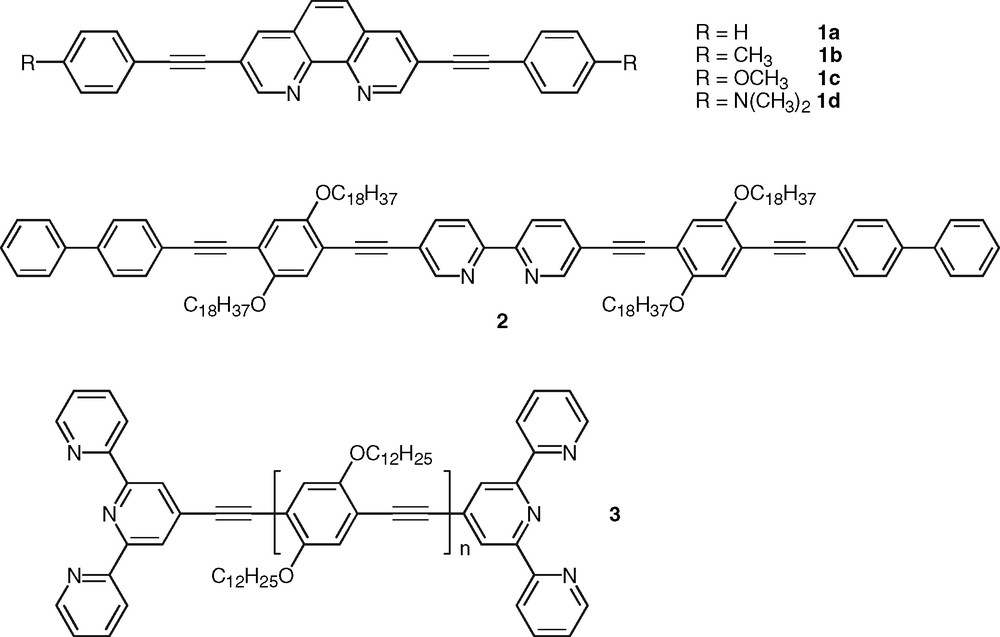
Selected examples of polypyridine ligands containing π-conjugated substituents.
Photophysical data for compound 1a–d in dichloromethane and acetonitrile as solvents (from [6])
| Compound | CH2Cl2 | CH3CN | ||||||
| λmaxa | λmaxb | λmaxc (Zn2+) | λmaxa | λmaxb | Φfd | τe | λmaxc (Zn2+) | |
| 1a | 346 | 376 | 415 | 342 | 375 | 0.41 | 2.93 | 434 |
| 1b | 350 | 384 | 436 | 346 | 384 | 1.0 | 1.50 | 453 |
| 1c | 358 | 416 | 490 | 350 | 441 | 0.64 | 1.71 | 530 |
| 1d | 410 | 523 | 646 | 402 | 587 | – | – | q |
a Absorption maxima (nm).
b emission maxima (nm).
c emission maxima (nm) in the presence of excess ZnCl2.
d fluorescence quantum yield.
e fluorescence lifetime (ns). –, not determined; q, quenched emission.
This approach based on the extension of delocalization using acetylenic linkage to favor highly allowed excited states with enhanced radiative transitions is quite general [23]. As an example poly(phenyleneethynylene)s constitute a very important class of photonic materials [24]. The same strategy was also proven successful in the case of bpy derivatives, such as compound 2 [25] that was synthesized as model of monodisperse oligomer for structurally-related bpy-containing polymers and investigated for the incorporation of transition metal ions within the conjugated backbone (Fig. 2). A series of soluble bis-tpy ligands, such as 3 [26], were also found to be highly fluorescent (Fig. 2). Compound 3 was designed as bridging ligand for the investigation of intermetallic energy transfer in binuclear metal complexes [15,27]. Polycyclic aromatic nuclei were also incorporated within conjugated ligands. These compounds possess low-lying singlet and triplet excited states and they can thus play the role of energy relay or antenna chromophore [28]. Particularly interesting are those conjugated polypyridine ligands that incorporate the pyrene nucleus owing to the many chemical and photophysical attributes of this polyaromatic group. Indeed, pyrene-containing molecules exhibit unique singlet state properties such as ability to form intramolecular charge-transfer states that are sensitive to environmental factors [29]. In line with our investigation on pyrene-containing receptors for transition-metal ions [30], we reported recently the synthesis and photophysical properties of pyrene–bpy dyads bridged by a phenylene diethynylene unit [31,32]. Compound 4 (Fig. 3) was shown to display intense fluorescence emission in the visible spectral domain that is not dependent on the solvent nature (Table 2), which stems from the weak electron-accepting property of the bpy ring (vide infra). This behavior is in contrast to the case of compounds 1a–d (Table 1) and particularly to that of dyad 5 in which bpy and pyrene units are held together in a linear arrangement via a single acetylenic bond [33] (Fig. 3). Indeed fluorescence in ligand 5 was observed to exhibit a strong solvatochromic behavior indicative of the formation of a polar excited-state, which was attributed to the additional electron-withdrawing effect of the acetylenic bond. This effect might be compensated to a significant extent by the electron-donating ability of the central di-alkoxy-phenylene unit in oligomer 4. The connection of the conjugated substituent at either the four- or five-position of the bpy unit was shown to have no significant impact on the photophysics of free ligands based on the phenylene ethynylene bridge [32]. Furthermore, the pyrene moiety was connected to the tpy unit via a diethynylated thiophene linker to give ligand 6 with a bent shape [34] (Fig. 3). Relative to the analog system 7 bearing a single triple bond [35], the introduction of the aromatic thiophene ring into the bridge has a major influence on the photophysical properties of 6. Indeed ligand 6 diplays fluorescence emission centered around 488 nm in acetonitrile, which was concluded to arise from a charge-transfer state. Clearly, this is not the case for 4, which underlines the importance of the nature of the bridge. It is also worth to mention the case of fluorene-based bpy ligands as oligo- or polyfluorenes represent promising materials for electroluminescene-based applications [36]. Recently, Ziessel and colleagues reported a modular approach toward the synthesis of a series of bpy-, phen-, and tpy-based molecules incorporating the bodipy chromophore. These systems are highly soluble and strongly fluorescent in organic solvents [37].
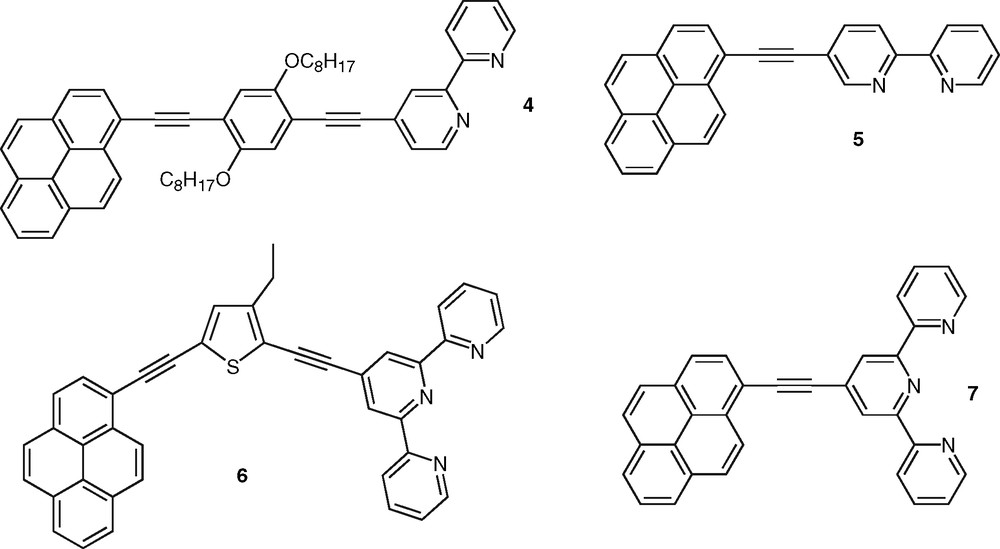
Pyrene–bpy ligands containing acetylenic bridges.
Photophysical data for compound 4 and its monuclear zinc(II) complex in toluene and tetrahydrofuran as solvents (from [32])
| Compound | Toluene | THF | ||||
| λmaxa | λmaxb | λmaxa | λmaxb | Φfd | τe | |
| 4 | 411 | 443 | 411 | 443 | 0.68 | 1.7 |
| 4-Zn2+c | 417 | 487 | 417 | 550 | 0.40 | 2.8 |
a Absorption maxima (nm).
b Emission maxima (nm).
c Data in the presence of excess ZnCl2.
d Fluorescence quantum yield.
e Fluorescence lifetime (ns).
Acetylene-based linkages clearly present appealing attributes for the construction of such conjugated structures and have been widely used for such purpose. They allow the generation of conjugated systems with well-defined geometrical shape and ensure efficient electronic communication along the molecular rods, which is due to the cylindrical symmetry of the triple bond. Highly fluorescent bpy ligands, such as compound 8, were also synthesized in which extension of conjugation is ensured by double bonds [38] (Fig. 4). The influence of the hybridization type of the conjugated linker on the efficiency of photoinduced intramolecular processes has been discussed [39]. Recently a non symmetrical phenylene-vinylene oligomer, 9, containing one terminal tpy unit was synthesized [40] (Fig. 4).
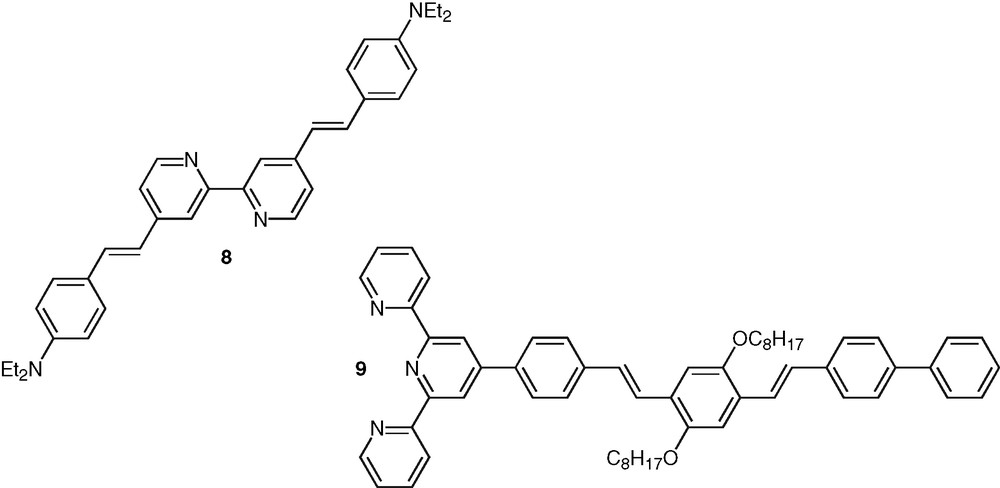
Selected examples of polypyridine systems based of vinylene spacers.
Finally, an important class of systems are those in which the polypyridine unit and an aromatic chromophore are connected by a single C–C bond. In contrast to the case of the ethynylated or ethenylated conjugated rods, the photophysical properties of the formers are very sensitive to the ground and excited-state geometrical properties. The torsion angle around the C–C bond plays a major role as it controls the extend of electron delocalization between the terminal units. Fluorescence emission in these systems is therefore quite sensitive to solvent polarity and arises from an excited-state with a more planar geometry than that of the ground state. This could be observed in the case of the bpy–pyrene ligands 10 and 11 [41,42] (Fig. 5) and confirmed with the study of a series of phen–pyrene compounds [43]. According to this feature, a bpy–pyrene derivative, 12, was proposed as model compound for fluorescent polarity probes [44] (Fig. 5). In the same connection, the substitution of bpy, phen, and tpy ligands with the 4-methoxyl-2,6-dimethyl-phenyl (or manisyl) motif offered access to a range of ligands, such as 13, 14, 15, respectively, displaying high fluorescence quantum yield and also high solubility [45] (Fig. 6). A tpy ligand containing the anthracene chromophore was shown to display a strong anthracene-type fluorescence, which is probably due to the orthogonal orientation of tpy and anthracene units caused by strong steric constraints [46].
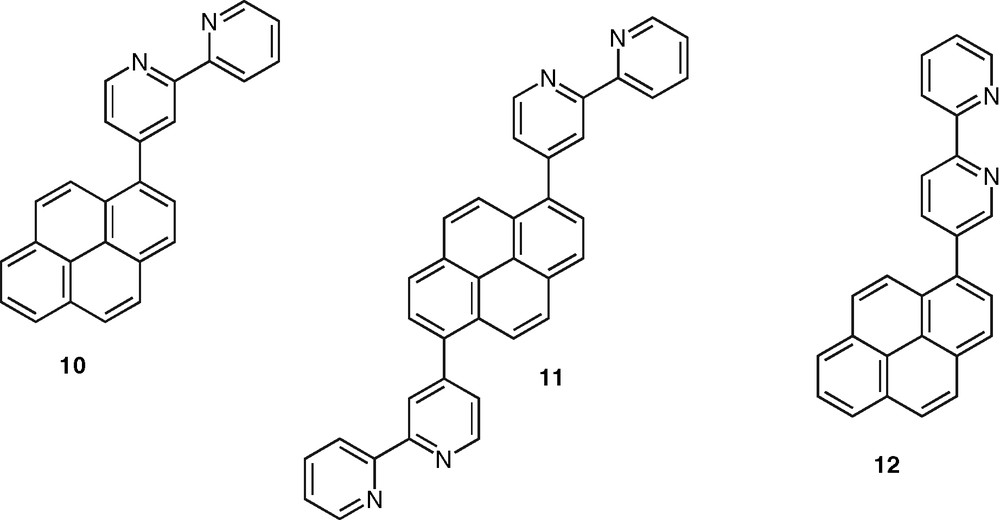
Pyrene–bpy ligands in which the aromatic hydrocarbon and heterocyclic moieties are connected via a single C–C bond.
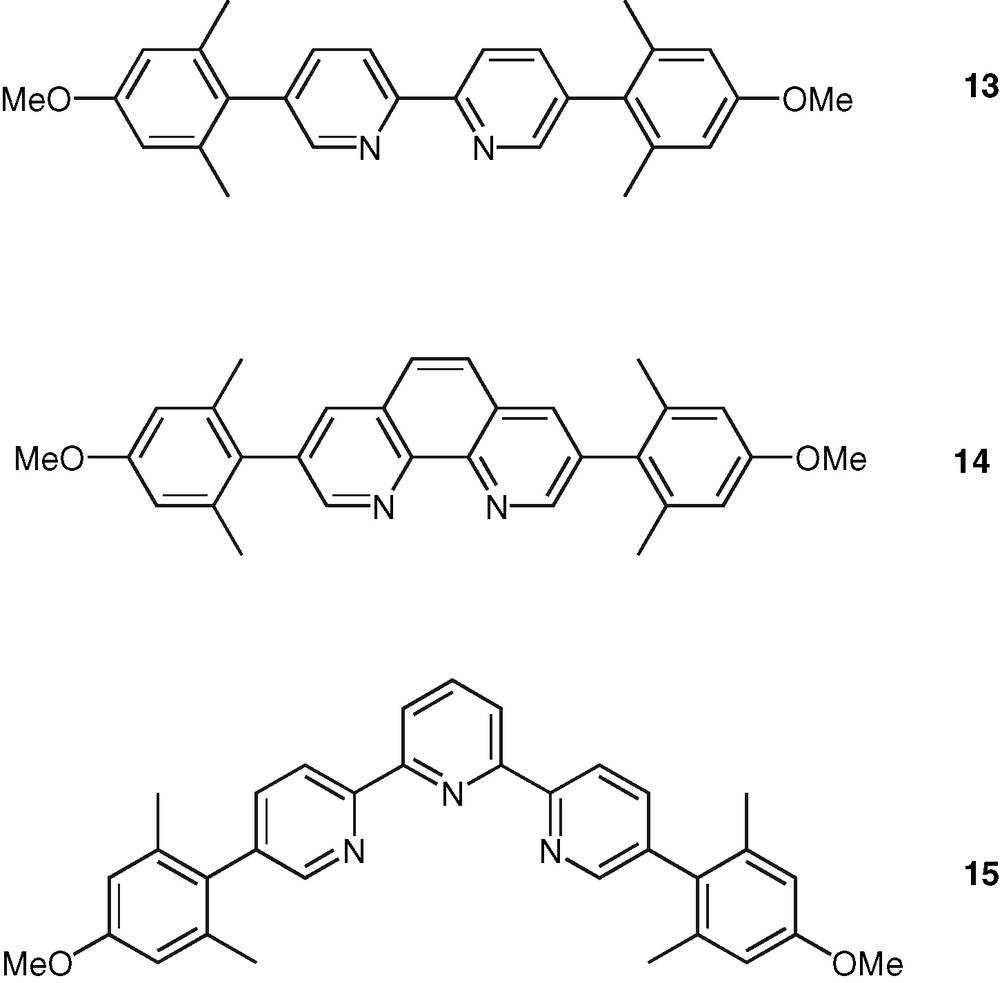
Examples of manisyl-substituted bpy, phen, and tpy molecules as tunable fluorophores.
3 Tuning the singlet excited states properties of conjugated polypyridine ligands
3.1 Effect of solvent polarity
Extending π electron delocalization in polypyridine ligands arguably provides a versatile means to generate bright fluorophores. As compared to the parent systems, emission is dramatically shifted toward the visible region and fluorescence quantum yields approach unity in certain cases. Solvent polarity can alter the photophysical features of the conjugated ligands but the effect is not general and strongly depends on the molecular structure. Bpy, phen, and tpy units are electron deficient yet not strong electron-withdrawing aromatic group. This is confirmed but the high reduction potential of these ligands which is usually found more negative than –1.5 V (vs. SCE) as measured by cyclic voltammetry, depending on the nature of the conjugative substituents [33,34,47]. This leads to small values for the exothermicity of the excited-state charge-transfer process [48]. As a result, the formation of polar ground and excited states in the π-conjugated ligands is therefore not favored, which is actually the case for compound 4 (Table 2). This is no longer true when strong electron-donating groups are attached, as exemplified by the phen-containing compound 3d, which contains the N,N-dimethylamino terminal group (Table 1) [6]. Torsional flexibility around the C–C bond in pyrene–bpy ligands can also induce a dependence of the Stokes' shift in solvents of different polarity, but the effect is rather small and the excited-state possesses a small extent of intramolecular charge-transfer [42,44].
3.2 Effect of metal-ion complexation
In contrast, protonation or metal ion complexation represent the most powerful way to alter the singlet state properties of polypyridine-containing conjugated systems. Particularly interesting is the fact that the dramatic photophysical changes are fully reversible. Indeed addition of a base or a competing ligand, respectively, totally restores the fluorescent features of the free ligand [6,32]. As mentioned above, charge-transfer phenomena play a central role in the photophysics of this class of molecular systems. Therefore, the effect of protons or metals is not surprising when one realizes that the attachment of such species to the ligand nitrogen atoms serve to increase the electron affinity of bpy, phen and tpy units to a considerable extent. For example, coordination of zinc(II) to the tpy moiety of compound 6 was shown to shift the tpy reduction potential to a much less negative value (–1.28 V vs. Ag/AgCl) [34]. As a result, the formation of highly polar excited states becomes favorable in the complex relative to the free ligand. The protonated/complexed species then behaves as a true push–pull chromophore (Fig. 7). This accounts for the considerable red-shift of fluorescence emission spectra upon protonation or ion binding. Indeed a 107-nm bathochromic shift of the fluorescence emission maximum was observed for the mononuclear zinc(II) complex of compound 4 in THF [31] relative to the free ligand (Table 2). As a consequence of the charge-transfer character of the emitting state, a strong solvatochromic effect is also observed when the fluorescence emission spectra of the zinc(II) complex are recorded in solvents of increasing polarity. The values of the excited-state dipole moments derived from these experiments are usually high, typically above 20 D, which confirms the occurrence of a giant dipole along the molecular axis. In highly polar solvents, such as acetonitrile, the addition of zinc(II) resulted in the quasi total quenching of the fluorescence emission and this can be rationalized in terms of the energy gap law. In simple words, the change in fluorescence emission maximum upon zinc(II) complexation corresponds to a change of the emission color and this has been observed for a variety of polypyridine ligands with extended conjugation, such as compounds 1a–d for examples [6]. The effect was also shown to be very sensitive on the nature of the complexed metal ion in the case of a series of 4,4′-bpy derivatives featuring donor/acceptor π-conjugated substituents [38]. These features endow the conjugated polypyridine ligands with interesting metal sensing properties [49]. Particularly, the conjugated ligands and their complexes display intense visible emission, as well as long excitation wavelengths, that render them promising molecular scaffolds on which to base the design of chromophores for biological investigations. Especially well-designed conjugated oligomers are of prime importance as chromophores displaying two-photon excited fluorescence [50]. Polypyridine conjugated ligands might thus represent promising systems in that respect. Another important feature is the strong dependence of the fluorescence emission properties of the metal complexes, especially the zinc(II) ones, on environmental factors such as the nature and polarity of the solvent. A straightforward application could be the use of such complexes in devices for the detection of volatile organic compounds [51]. Moreover, the fluorescence quenching that prevails in complexes of closed-shell metal ions in polar solvents or empty-shell metals, such as transition metal ions, corresponds to the ‘OFF’ situation, while the ‘ON’ signal is set by the intense fluorescence emission from the free ligand. This ‘ON/OFF’ modulation of the optical signal is critical not only for sensing purposes, but also for the generation of logic gates performing molecular-scale arithmetic [52]. Finally, zinc(II)-induced formation of charge-transfer states in conjugated bpy ligands has been elegantly exploited for the generation of functional complexes with nonlinear optical activity [53,54].
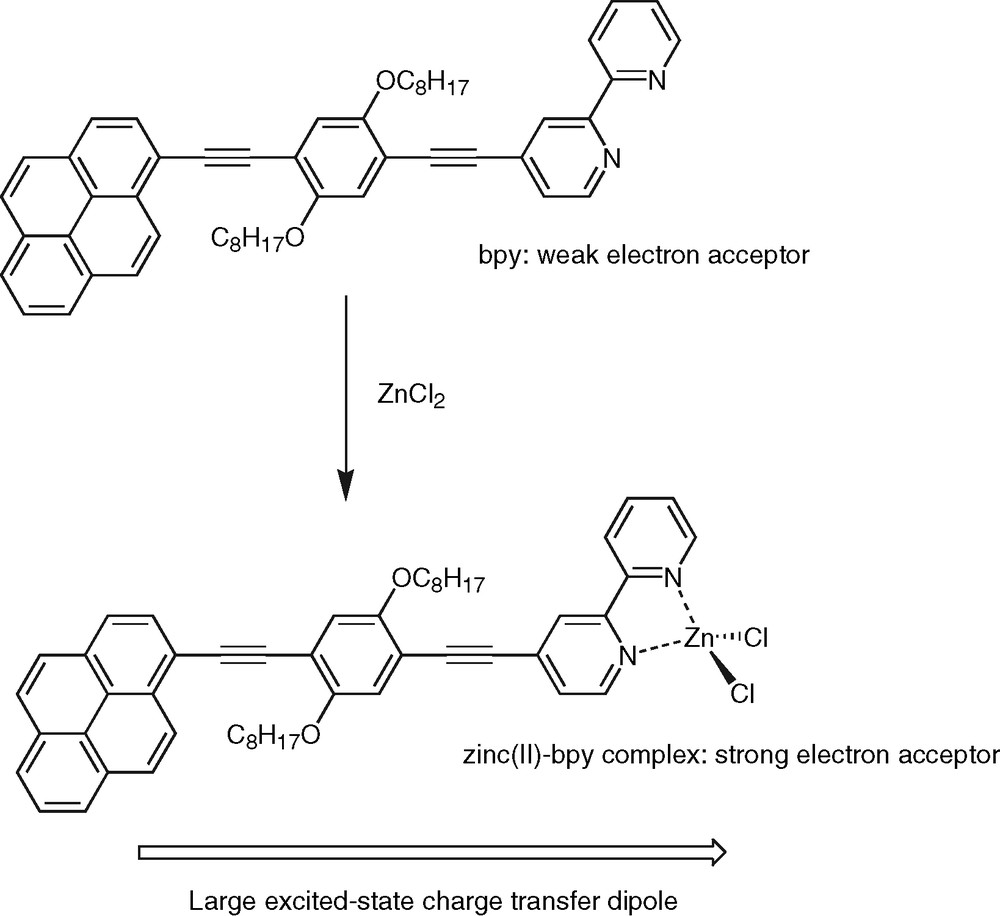
Zinc(II)-induced formation of a charge-transfer excited state within a polypyridine conjugated ligand.
4 Conclusion
This account was intended to outline the potential of conjugated polypyridine ligands as versatile fluorophores with improved fluorescent properties. As compared to the parent bpy, phen, and tpy molecules, the enhanced fluorescence emission in the π-conjugated derivatives stems from the extension of electron delocalization over the molecular skeleton, which induces a substantial stabilization of the fully allowed π−π* states and thereby an increase of the radiative deactivation efficiency. Particularly attractive is the ability of such systems to undergo profound photophysical changes upon metal complexation owing to the inherent coordination properties of the bpy, phen and tpy units. From the photophysical standpoint, this behavior is related to remarkable ability of the conjugated oligomers containing polypyridine ligands to undergo photo-induced charge-transfer in solution upon introduction of exogenous additives (protons, metal ions). Ready tunability of excited-state properties turns out to be a very important functional feature for the incorporation of the conjugated ligands into materials and more sophisticated architectures. While focus was given therein on low molecular weight oligomers, examples of the macromolecular counterparts have been reported as well. They combine the uniques attributes of conjugated polypyridine structures with the processability of polymers. In that respect, they allow to produce a variety of materials for many applications, such as metal ion chemosensing [7], electroluminescence [55], liquid crystals [56], or photoconductivity [57]. Conjugated polypyridine ligands are also being exploited as building-blocks for the constructions of nanostructures such as macrocycles [58] [59], hyperbranched structures [60], polytopic modules for nanoengineering [61], or self-assembled chromogenic ion sensors [62]. Moreover, this account emphasizes the behavior in the singlet excited-state but conjugated polypyridine ligands also allow to investigate triplet state properties. Particularly ruthenium(II) complexes display unique luminescence properties [12] that are markedly affected by conjugative substituents. Very long-lived excited states can be generated, especially in the case of pyrene-containing ligands [63]. π-Conjugated polypyridine ligand arguably represent an important class of versatile ligands that offer attractive photophysical and coordination features for the generation of a wide range of molecular and supramolecular structures and materials.