

1 Introduction
Of the elements essential to life, metals compose an important fraction [1]. Thus, coordination chemistry, in the sense that it is very largely the chemistry of metal cations, underlies many of the most fundamental of biological processes. With certain spectacular exceptions, in particular for the haem proteins haemoglobin and myoglobin [2], however, most knowledge of the precise functions of metal ions in biological systems remained diffuse and speculative for at least fifty years after Alfred Werner’s rationalisation of the nature of metal complex compounds1. In some instances, the enzyme urease providing a well-known example [3], even the presence of a metal ion in a familiar biomolecule remained unrecognised. From the 1950s on, this situation began to change rapidly in response to many stimuli resulting from advances in both chemistry and biology, one of the most significant of these early advances being the development by Manfred Eigen of methods for the study and interpretation of very fast reactions of metal complexes [4]. An expression of the resulting closer interactions between chemists and biochemists interested in the behaviour of metal ions was the definition of a domain of ‘bioinorganic chemistry’2. The need to understand the antibiotic action of naturally occurring materials, ultimately identified as ‘ionophores’ [6], (Fig. 1) involved in the transport of sodium and potassium across cell membranes can be seen as one of the bases for the development of the chemistry of synthetic ion transporting agents recognised with the award of the Nobel Prize in 1987 [7], and these synthetic materials in turn have had their benefits to medicine [8], as have many other metal chelating agents [9]. Rationalisation of the coordination of main-group cations such as Na+ and K+, frequently still discussed in terms of ‘electrostatic’ or ‘ion–dipole’ interactions, is an area of continuing concept development, stimulated further by observations indicating the importance of polyhapto binding of Cs+ to reagents used in the treatment of nuclear waste [10]. As another stimulus to both chemistry and biology, early efforts to understand the bactericidal action of simple metal complexes of aza-aromatic ligands [11] can be seen as a foundation to now extraordinarily extensive investigations of the interactions of metal complexes with DNA [12] including, for example, study of the use of platinum complexes as anti-cancer agents [13]. Many other expressions of the mutually beneficial interactions between chemistry, biochemistry and medicine can now be found, one that is particularly well known and at a high level of sophistication being the development of relaxivity enhancement agents for magnetic resonance imaging (MRI) based on macrocycle complexes of gadolinium(III) [14]. Comprehensive discussions of the remarkable development and continuing vigour of the field of bioinorganic chemistry can be found in special editions of the journal Chemical Reviews devoted to ‘Medicinal Inorganic Chemistry’ [9], ‘Bioinorganic Enzymology’ [15a] and ‘Biomimetic Inorganic Chemistry’ [15b]

Views [16] of the structure of the K+ complex of the macrocyclic ionophore valinomycin [6]. Shown in red are the carbonyl-O atoms of the ester units and water-O atoms, considered to be the ligand donor atoms. Note that bonds to the metal are not shown, perhaps a reflection of a particular view of the interaction as an ‘ion–dipole’ attraction that should not be indicated by line connecting the centres ?
In the early phase of the development of bioinorganic chemistry, a strong focus was upon the study of simple complexes as ‘models’ of the active sites in numerous enzymes and other metalloproteins3. In some cases, such as attempts to mimic the behaviour of iron–sulphur proteins, the synthetic complexes provided extremely close models of the protein-bound centres [18]. In others, such as attempts to understand the activation of nitrogen by nitrogenase, it led to a flowering of a major field of metal/nitrogen coordination chemistry [19], though not to the definition of a precise mechanism for the enzyme action. Lying between such extremes, many studies of models for enzymes such as peptidases [20], phosphatases [21] and dehydratases [22] led to useful analyses of the factors underlying enzymic activation of substrates and provided an enormous stimulus to the field of ‘reactions of coordinated ligands’ [23], an area obviously of relevance also to abiological catalysis. Important results here were the recognition of the importance of solvation changes induced by coordination (as distinct from ‘polarisation’ effects within the metal–ligand bond) [24] and of the efficacity of the metal in rendering an intermolecular process intramolecular [25]. The success of some of these models was perhaps largely due to the relative simplicity of the enzyme’s active site and the availability of a small number of crystal structures [26] which enabled the coordination sphere of the catalytic metal to be established with some certainty. The revolution in crystallographic techniques and, in particular, the rise of protein crystallography which characterised the last quarter of the twentieth century, however, have dramatically modified perspectives in bioinorganic chemistry and provided some fascinating problems in understanding the roles of metal ions in multimetallic systems. This is illustrated presently by consideration of some of the more spectacular results and truly extraordinary achievements of metalloprotein crystallography, the structure solutions for nitrogenase [27], cytochrome-c oxidase [28], and the reaction centre [29] and oxygen-evolving centre [30] of the photosynthetic apparatus. A particularly thorough review of structural bioinorganic chemistry [31] given some years ago provides a comprehensive background to these remarks. Figures drawn to illustrate the following discussion are based upon coordinates available from the Brookhaven Protein Data Bank (PDB) [32], also a source of much more comprehensive biochemical information than is presented herein.
2 Coordination chemistry in multimetallic biosystems
2.1 Nitrogenase [27]
Reduction of a structure as elaborate as that shown for nitrogenase from azotobacter vinelandii (Fig. 2) to those of the metal-containing units with their primary coordination sphere ligands only provides a first step in analysing the coordination chemistry involved. Since nitrogenase is a reaction catalyst (for the reduction of nitrogen to ammonia), a full understanding of the functions of the metals present must ultimately go beyond analysis of the structure of the enzyme, though this is a difficult step still fraught with pitfalls. At the structural level, three distinct complex entities can be identified. One of these is a ferredoxin unit of Fe4S4(SR)4 stoichiometry, an example of one of the first-recognised polymetallic biological coordination complexes [18] and commonly considered to exemplify a primitive means of solubilising the versatile transition metal, iron. A conventional representation of the structure is shown in Fig. 3 (left) and, as is almost always true for coordination complexes, a single line between a metal and a donor atom is used to denote a bond that may well be something quite different to a 2-centre/2-electron interaction. In iron/sulphur protein clusters, this is not simply because both sulphide and thiolate ligands may act as π- as well as σ-donors but because the electron density within the cluster is not localised, so that the description of a species such as [Fe4S4(SR)4]2–, for example, as containing 2 Fe(III) + 2 Fe(II) is a formalism [18]. The space-filling representation in Fig. 3 (right) involves no bond-line ambiguities but simply exchanges this for uncertainty as to what is indicated by a van der Waals radius and how transferable such parameters may be from one situation to another.
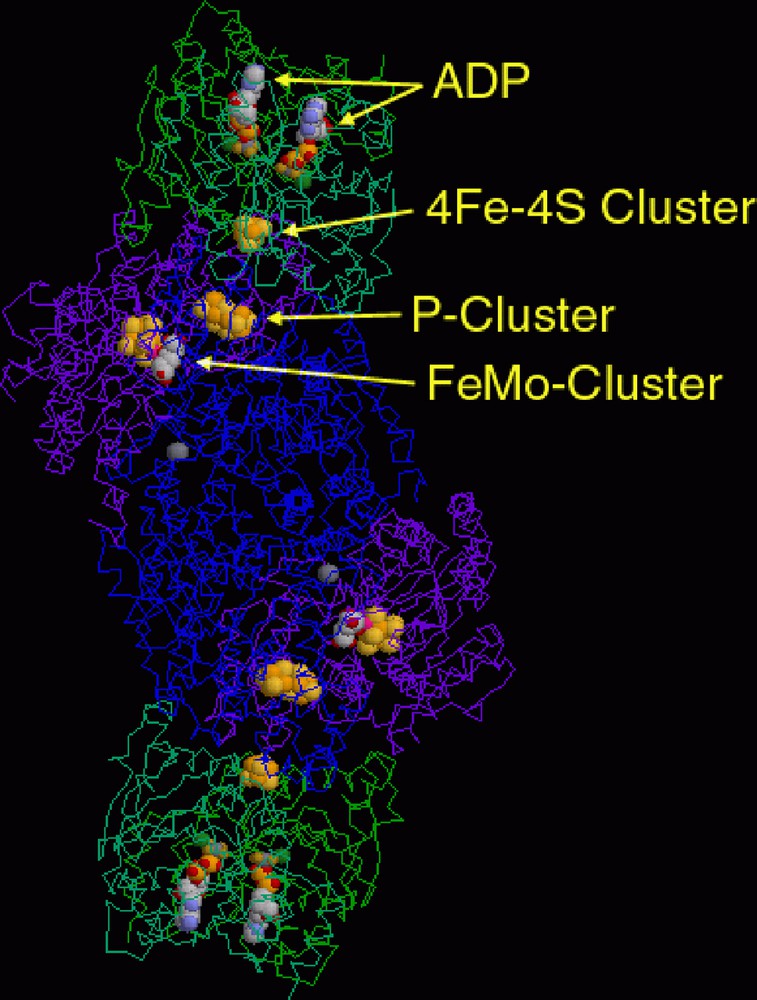
A representation of the dimeric nitrogenase protein from azotobacter vinelandii (PDB code 1N2C), showing the positioning of the various redox-active metal complex units.

Ball-and-stick and space-filling representations of the ferredoxin unit present in nitrogenase.
There is clearly a need and one not just limited to biological coordination complexes, to find better means of representing the precise nature of a coordinate bond. It is one of the interesting differences in convention between ‘organic’ and ‘inorganic’ chemistry that the formula for an unsaturated carbon compound would be expected to show a multiple bond but that a formula for a ‘coordinately unsaturated’ transition metal complex would almost never indicate more than a single bond between the metal and a donor atom. The iron atoms in ferredoxins can, of course, be regarded as coordinately unsaturated in the sense that they are only four-coordinate and if each bound sulphur is considered to donate two electrons, then only eight electrons are added to a valence shell containing either five (Fe(III)) or six (Fe(II) electrons and the Kr configuration is not attained. Such non-conformity to the ‘Effective Atomic Number’ (EAN) Rule is not an observation unique to biological systems, but its recognition there does provoke the question of what factors limit the coordination numbers of metal cations, a question which is highly relevant to biocatalysis, since frequently it appears necessary to postulate changes in coordination number during a catalytic cycle [33]. In terms of simple molecular orbital theory, both the Octet and EAN rules can be seen as interdictions of the occupancy of strongly antibonding MOs and not as requirements that the coordination number consistent with this limit be attained. If the Fe–S bond is considered to be of an order higher than 1, the problem disappears, though it is then useful to note that addition across such a multiple bond could occur to increase the coordination number of Fe while it would still conform to the EAN rule. These, of course, are issues concerning the primary coordination sphere of the metal but one of the features of metalloenzyme systems, where the metal is often buried deep within the protein structure, is the influence of secondary and higher coordination sphere components upon the properties that the presence of the metal endows upon the protein. The iron/sulphur proteins show, par excellence, this form of sensitivity, their redox potentials for formally identical changes of oxidation state varying widely as a function of the particular protein environment [18]. Protein crystallography can be regarded as a unique means of precisely defining coordination spheres well beyond the primary that should persist in solution, whereas for simple metal complexes in solution, definition of even the secondary coordination sphere is generally extremely difficult [34]. Factors such as ‘allosteric’ [35] and ‘entatic’ [36] effects, undoubtedly important in all chemistry as a means of understanding how a molecule may be considered prepared for reaction, find their most precise definition in biological examples. The ferredoxin unit of nitrogenase is considered to be the first unit of the complete protein to undergo the electron acceptance which eventually leads to the reduction of N2 and its position, bridging two protein subunits, is relatively exposed to solvent access, leading thus to an environment and electrochemical behaviour unlike those of similar clusters in other proteins. This bridging of protein subunits is also illustrative of the way coordination may have a long range influence on structure in elaborate systems, and is seen again in the case of the photosynthetic reaction centre [29], for example (see below).
The other polymetallic units of nitrogenase, the ‘P-cluster’ and ‘Fe-Mo co-factor’ (Fig. 4) were, at the time of their initial characterisation, of unprecedented forms and their discovery stimulated an extensive range of complex ion synthesis providing many simple analogues [31,37]. Interestingly, refinement of the nitrogenase structure [38] has shown that a light atom (probably N) was omitted from the original FeMo-co unit, indicating that even a partly garbled signal can produce a useful response! Both the P-cluster and FeMo-co must have redox functions, with the P-cluster accepting electrons from the Fe4S4 cluster before donating them to FeMo-co, and this complexity of electron-transfer pathways that typifies so many biological processes presumably reflects the need for efficiency, gained by transferring an electron in several steps with small individual reorganisational barriers rather than in a single step likely to have a large reorganisational barrier [39]. Nonetheless, differences in structure of reduced and oxidised forms of the P-cluster are readily detected crystallographically [27,37,38]. The very observation of rapid, ‘long-range’ electron transfer between biological redox entities typically 10–15 Å (if not further) apart, as in nitrogenase, has been a great stimulus to the development of improved theories of electron transfer and a better understanding of conductivity in molecular materials [40].
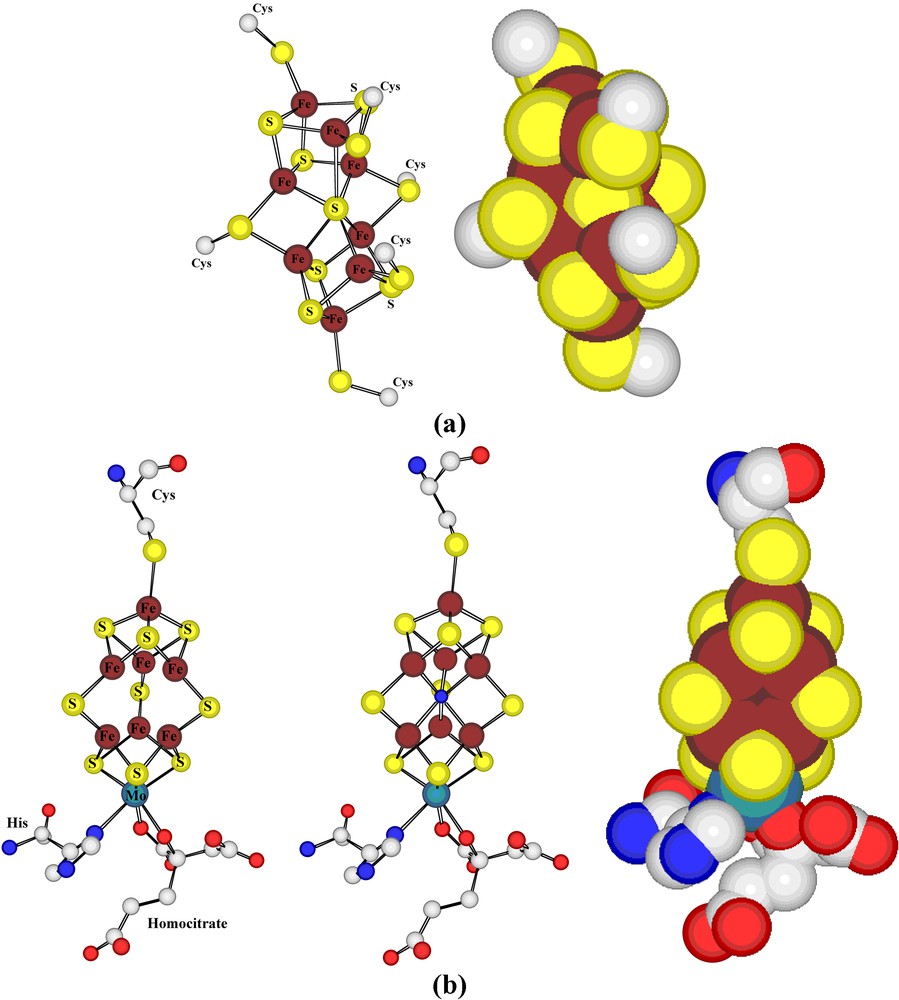
(a) Representations of the dithionite-reduced form of the ‘P-cluster’ of nitrogenase, a kind of fused ferredoxin species. (b) Views of the dithionite-reduced form of the ‘FeMo-cofactor’ of nitrogenase: (left) as defined in the initial structural study; (center) as modified following later structural refinements to include a central light atom (here shown as blue for N); (right) as a space-filling form of the latter, showing how the central atom is encapsulated by the cluster.
Though it is clear that both the P-cluster and the FeMo-co unit must be involved in electron transfer processes, one of the oft-remarked aspects of the determination of the nitrogenase structure is that it did not provide an obvious explanation of exactly how the complicated process of a six-electron reduction of N2 might occur (despite long-held hopes that it would) [19,27,33,37,38]. Here, some important aspect of coordination remains well hidden! One of the remarkable features of all iron-sulphur protein chemistry is that, despite the evidence for the importance of coordinated nucleophile activity in enzymes such as carbonic anhydrase [17b,26] and the general knowledge that a coordinated nucleophile may retain a vastly greater fraction of its nucleophilicity than it does of its basicity [41], there are no indications that coordinated sulphide or thiolate of iron–sulphur proteins have any direct role in the enzymic processes (even that of aconitase, a well-known case where redox activity does not appear to be relevant [42]4). Perhaps, given also that coordination of N2 could enhance its electrophilicity and that the close positioning of N2 and S in the coordination sphere of one metal ion could accelerate their reaction, nitrogenase is the system where the nucleophilicity of bound sulphide/thiolate might finally be revealed to have a biological role? Of course, if it correct that a nitride ion is the species surrounded by six Fe atoms in FeMo-co, perhaps it, too, could have a role as a nucleophile to add to and so activate N2, though it is particularly clear in the space-filling view of the reduced form of FeMo-co (Fig. 4) that this atom is well protected from its environment. Regardless of such speculations, nitrogenase and its properties continue to provide provocation for a better understanding of coordination and its consequences.
2.2 Cytochrome-c oxidase [28]
Were it only to be considered for the number of metals present and the variety of their binding sites, this enzyme would still be remarkable! It contains binding sites for Na/Ca (depending on the source and oxidation state5), Mg, Fe (× 2), Cu (× 3) and Zn, illustrating nicely how biological systems may be considered to exploit the coordination of metals of all kinds. The redox-active metals, Fe and Cu, are those considered essential for the electron-transfer capacity (involving direct conversion of O2 to H2O) of the enzyme but the properties of these centres are clearly regulated by structural factors which are at least partly influenced by the other metals [28a,43]. Once more, ‘coordination’ must be considered something that involves significant interactions well outside the primary coordination spheres of the metals. Though the immediate environments of all the metals in cytochrome-c oxidase are unexceptional (presuming that for Na/Ca and Mg, apparent coordination site vacancies are occupied by water molecules) given what is known of their general coordination chemistry, this is another biosystem that beautifully illustrates the subtle control of redox properties given by the appropriate choice of donor atoms. Aqueous solutions of simple Cu(II) compounds will oxidise thiols to disulfides but there is a dicopper site in cytochrome-c oxidase in which the copper atoms are bridged by two cysteine-thiolate donor atoms that remains intact when the enzyme cycles through a Cu(II) form. Protein-sidechain donor atoms from histidine (N), alanine-carbonyl (O) and methionine (S) appear to provide the necessary degree of stabilisation for this to occur (parallel to the situations for a number of other Cu enzymes [5d,44]) (Fig. 5).
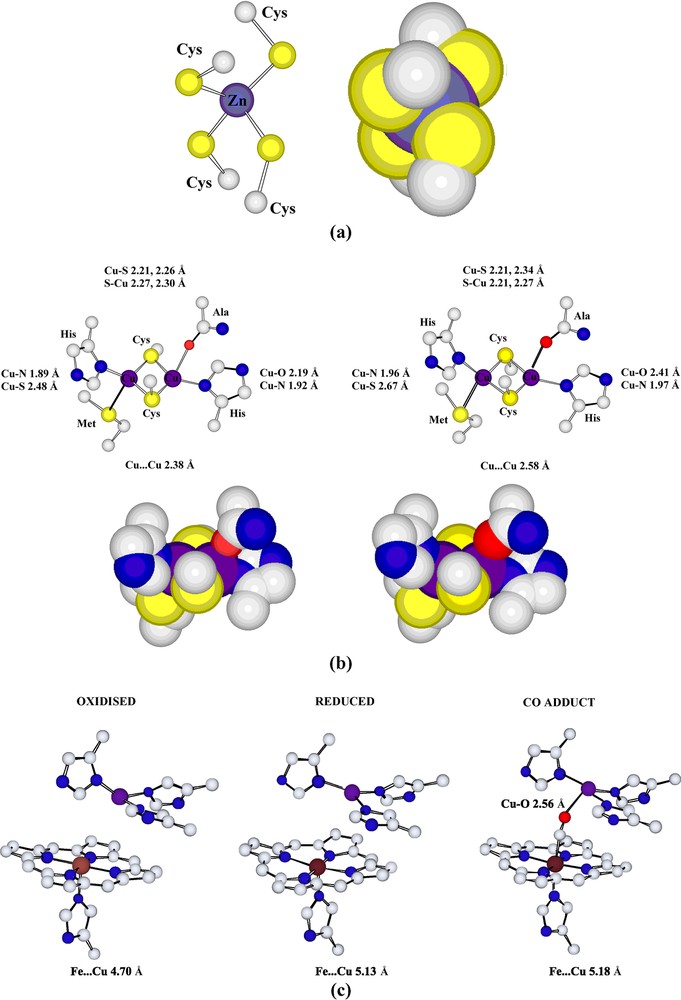
(a) The tetrahedral structural [Zn(Cys)4]2– unit of cytochrome-c oxidase, which is of closely similar form in both oxidised and reduced forms of the protein. (b) The cysteinate-bridged dimeric copper unit of cytochrome-c oxidase in the fully oxidised form (left) and the fully reduced form (right) of the protein. (c) The oxygen binding/reducing Cu–Fe site of cytochrome-c oxidase in the fully oxidised (left), the fully reduced (centre) and CO-bound (right) forms of the protein. (PDB codes 1OCC, 1OCR and 1OCO for the bovine heart muscle oxidase.) Both porphyrin and amino acid entities are shown to be truncated.
The site of O2 reduction in cytochrome-c oxidase is considered to be the Cu–Fe entity in which the Fe is bound to a porphyrin and the Cu to three histidine imidazole side chains, the structure of the CO adduct of the enzyme (Fig. 5) presumably indicating the way in which O2 may be bound between the two metals. Reductions of fundamental importance such as that of O2 to H2O and N2 to NH3 are, of course, multi-electron processes that may involve the formation of various stable intermediates, and the ‘sandwiching’ of O2 between two metals as in cytochrome-c oxidase can be seen as a way of trapping both the substrate and intermediate reduction products for long enough for the overall reduction to be efficient [45]. The idea of creating a site suitable in particular for N2 to be trapped by two metals was the basis for a major interest in the synthesis of ‘binucleating ligands’ [46] and thus an important impetus for the development of a significant new field of coordination chemistry [47].
3 The photosynthetic reaction centre [29,39]
A somewhat extraordinary fact is that the metal present at the centre of the photoredox apparatus that may be considered one of the bases of life on Earth is the redox-inactive species Mg(II). Whatever influence Mg has upon the properties of the ‘special pair’ of chlorophyll molecules within which the initial photo-induced charge separation, the beginning of photosynthesis, occurs, it must be a more subtle effect of coordination than to simply offer a site for electron transfer. Since in the protein constituting the photosynthetic reaction centre there is an array (Fig. 6) of metals (and macrocycles) including a porphyrin bound, redox-active metal (Fe) involved in electron transfer following the initial charge separation, it is clearly not necessarily disadvantageous generally for a redox-active metal to be present, though it is also interesting to note that the non-haem Fe present appears to have a structural function only. If the involvement of a pair of chlorophyll molecules as the reaction initiator signifies the fact that a large distance of charge separation is necessary for redox reactions to compete significantly with charge return [39], then perhaps the role of Mg is to control the degree of association between the two molecules. There is some degree of π-stacking [48] in the special pair (Fig. 7) and this is an effect that, in numerous simple complexes of aromatic ligands, is seen to be sensitive to the particular metal involved [49–51]. Again, biology may be indicating that coordination is an event with major consequences that extend well beyond the atoms in closest proximity, though to identify the cause of an ostensibly weak interaction is always difficult and it may be that long Mg···O contacts between the chlorophyll molecules are also important.

The marvellous array of metals and macrocycles buried within the protein of the photosynthetic reaction centre. (Peripheral substituents on the macrocycles are not shown.).

Views of the ‘special pair’ of chlorophyll molecules within the photosynthetic reaction centre: (left) A perspective view showing the bound histidine side chains; (right) a simplified view, with all atoms of a given Mg-macrocycle unit of one colour, perpendicular to the mean macrocycle ring plane and showing some of the close atomic contacts.
4 The oxygen-evolving centre (OEC) [30,52]
As another vital component of the photosynthetic apparatus, the OEC has been the subject of intensive study and many aspects of the structure finally revealed by crystallography, for example, the presence of four Mn atoms at the active site, had been accurately predicted from spectroscopic measurements coupled to extensive ‘modelling’ based on syntheses of a wide range of polynuclear Mn complexes [52]. However, although oxo-bridged, cubane structures for this Mn4 unit had been suggested [53] (and discounted [52]), the cubane species found in the crystal structure [30] (Fig. 8) contains only three Mn atoms in the cube, with a fourth being pendent and the fourth metal atom in the cube being Ca. The ‘external’ ligands to the cube appear to be carboxylate-O (aspartate, glutamate), bicarbonate-O (to Ca only) and histidine imidazole-N, an environment that raises issues both in regard to the accessibility of oxidation states and light absorption by the cluster [54]. As a multidentate ligand, a protein offers a variety of modes for controlling the spatial distribution of side-chain donor atoms [55] and one obvious inference from the OEC structure is that synthetic ligands might well be designed to provide specific isomeric forms of small mixed-metal cluster units. Given the importance of metal cluster chemistry, this may well be another useful stimulus.

Views of the Mn4CaO4 cluster present in the photosynthetic oxygen-evolving centre, with and without ligating groups from carbonate and amino acid side chains.
5 Conclusions
The metalloproteins discussed above are but a few examples of the biological systems in which metals are believed to have a central function. There is no reason to consider that any distinction of biological coordination chemistry from coordination chemistry in general is anything other than a convenience for its description, and there are innumerable examples of biological coordination that are very simply rationalised in terms of models as venerable as that of Alfred Werner. What studies of many biosystems have shown is the difficulty involved in providing a precise quantitative interpretation of ‘coordination’, which in the simplest sense means no more than that two entities have ‘similar’ Cartesian coordinates, and its consequences. These consequences may be structural, electronic and kinetic, and the last of these, the basis of enzymic catalysis, is certainly not yet one that is fully understood in any domain of coordination chemistry.
Finally, it may be noted that ‘coordination’ in biology need not be associated with the presence of a simple metal cation. Recently, great attention has been focussed on ‘cation-π’ interactions involving quaternary ammonium cations [56,57] and that may control, for example, the docking of acetylcholine in acetylcholine-esterase [58]. The question of the precise origin of such interactions is an interesting one but it may be yet another reflection of the remarkable coordination chemistry of the proton, a chemistry much better known under the title of ‘hydrogen bonding’ [59]. It is also reflected in chemistry such as ‘bridge bonding’ in boranes [60] and ‘agostic’ interactions of transition metal complexes [61], and once again it is possible that biology may eventually provide the stimulus to reconcile seemingly disparate aspects of the chemistry of a given element.
1 Note that even the intense scrutiny given to these proteins did not serve to define all possible modes of Fe/O2 interaction, see [2c].
2 A selection of texts that illustrate both the nature and the development of this field is given as reference [5].
3 Discussed in the Editorial Comments in references [14]. For thorough reviews of some very sophisticated modelling studies, see [17]. See also references [18,19].
4 Note, however, that there is evidence for the importance of bound sulphide nucleophilicity in precatalytic steps for some hydrogenases, see [42b].
5 See PDB entries 1OCC, 1OCO, 1OCR and 1AR1, for example.