

1 Introduction
During the last decade, the domain of inorganic chemistry enriched of sophisticated objects showing high levels of complexity in composition, geometry and properties. This tendency results from the development of new concepts in inorganic chemistry, namely supramolecularity, host–guest chemistry, building block approach, all these concepts owing much to the recent improvements in structural characterization techniques. Now, XRD and solid-state NMR methods make possible the complete characterization of giant molecular species up to the nanometer scale. Most of these compounds are deliberately prepared in aqueous solution, sometimes in organic solvents, and are recovered by crystallization (single crystals) or by precipitation (powder). As the size of the soluble species is increased, the problem of the solvent–compound interactions becomes crucial. Indeed, to avoid the self-aggregation or spontaneous precipitation from solution, it is necessary that these large molecules adapt their size and shape in solution in order to minimize their interactions, a complementary way being to tune their contact surface with the solvent to idealize the entropy value of solvent-interaction. The molecular surfaces that mostly achieve these two ideas are that of balls and rings. Probably, the most spectacular reported examples are those developed by A. Müller, see for example the soluble ‘hedgehog’ [1] which contains 368 atoms of molybdenum. Another representative example is that of polyoxometalates (POMs), a family rich of thousands examples of nanometric balls used in catalysis, medicine, material chemistry, analytical chemistry [2]. POMs result from acidobasic condensation processes of soluble oxo-metalates [MO4]n– (M = V, Mo, W, Ta, and others), the junctions between the metallic centers being ensured by oxo- and/or hydroxo-bridges. We took advantage of our experience in POM chemistry and condensation processes to design cyclic architectures far from the POM archetypal geometry but prepared through similar acidobasic condensation reactions. To achieve this purpose, which means to design soluble cyclic architectures susceptible to be selectively isolated in the solid state, we were led to imagine new precursors that could be used in the building block approach concept. Referring to the topic of this special issue, this contribution is limited to some of our recent examples of cyclic clusters derived from precursors containing a metal–metal bond.
2 Precursors and reaction conditions
The design of sophisticated clusters by the building block approach requires specific precursors since the properties of the step by step resulting cluster closely depend on that of the precursor. A good precursor is expected to display a reasonable thermodynamic stability along the condensation process, an adapted electronic structure is required, and its initial geometry has to be chosen with respect to that of the targeted product. According to previous works in Mo/O/S chemistry developed in Versailles, the dications {MoO2S2}2+ and {Mo2O4}2+ were chosen for matching these requirements. The two precursors have the same overall molecular structure; they differ only by two bridging sulfur atoms replacing the two corresponding oxygen atoms. The geometry of {MoO2E2}2+ is represented in Fig. 1.
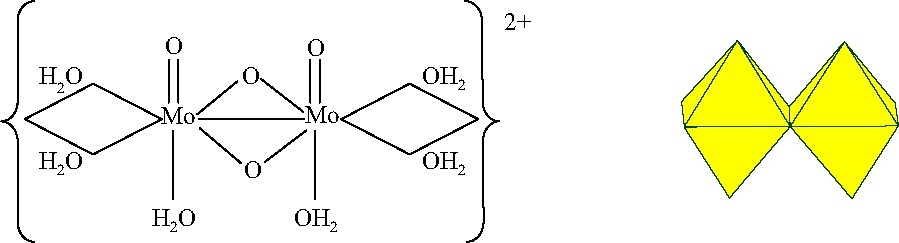
Representations of the {Mo2O2E2}2+ building blocks with the Mo–Mo metal bond.
The two equivalent octahedral MoV (d1) centers are mutually linked by a double E-bridge (E = O, S) each Mo atom being bonded to a terminal oxygen through a short multiple bond (1.64 Å). The two Mo = O fragments occupy equivalent syn positions. The two d1electrons of the Mo atoms are coupled to form a metal–metal bond. The Mo–Mo distance is very short, 2.6 Å for E = O and 2.8 Å for E = S (for comparison MoVI–MoVI distances are about 3.5 Å). This metal–metal bond increases the stability of the dication (stable in HCl 12 M). In aqueous solution, three water molecules complete the coordination of each molybdenum atom to form the aqua dication [Mo2O2E2(H2O)6]2+. The three water molecules are not equivalent since the Mo–OH2 bond trans to the Mo = O double bond (axial position) is weaker than the two others (equatorial positions) as revealed bond lengths and reactivity. Each Mo atom is highly charged (+5) that induces strong polarizing effects to the terminal water ligands, which are polarized enough to be ionized. Indeed, inter-building block connections via a Mo–OH–Mo bonding scheme can be established by action of a base in the presence or not of an assembling group (AG). The object of the paper is limited to the last point.
| n [Mo2O2E2 (H2O)6]2+ + m OH– + AG → polyoxo(thio)metalates |
The condensation process is controlled by the usual factors in this matter, concentration, reagents stoichiometry, pH, and ionic strength. The choice of the geometry of the assembling group is a key point since it permits to direct the molecular arrangement and the solid-state dimensionality. Some examples of different AG expected to orientate the condensation of the oxo and oxothio precursors are examined.
2.1 [Mo2O2O2(H2O)6]2+
The MoV-oxodication was obtained by reduction of a MoVI-precursor, generally commercial Na2MoO4·2 H2O. Depending on the synthesis conditions, two reducing agents were readily used, metallic molybdenum for hydrothermal syntheses, hydrazine for standard solution reactions at room temperature [3]. Molecular species were prepared by standard acidobasic condensation methods while one-pot hydrothermal techniques were reserved to generate solid-state materials (these solids are generally insoluble).
2.2 [Mo2O2S2(H2O)6]2+
The oxothio precursor was obtained by the selective oxidation of the terminal disulfure ligands in the [(S2)MoOS2MoO(S2)]2– anion by iodine. This protocol initially described for DMF medium [4] was adapted by us to aqueous solutions [5]. The use of water as solvent is really important since it permits the control of the condensation by simple pH measurements.
3 [Mo2O2O2(H2O)6]2+ based wheels
3.1 Phosphates as assembling groups in hydrothermal conditions
Molybdenum phosphates containing transition metals are interesting species for combining the properties of polyoxometalates and those of the metal phosphate (magnetic properties, microporosity) [6]. In the chemistry of MoV and phosphates, the very stable [P4Mo6O31–x(OH)x]12–x series is quasi systematically obtained, the degree of protonation varying from one structure to another [7]. In all these structures, see Fig. 2, a peripheral phosphate and a hydroxo group mutually connect the {Mo2O4} units.
Representations in ball and sticks and polyhedra of [P4Mo6O28(OH)3]9–, and of the dimeric [M(P4Mo6O28(OH)3)2]16–, M = Na, Mo are orange, PO4 are green.
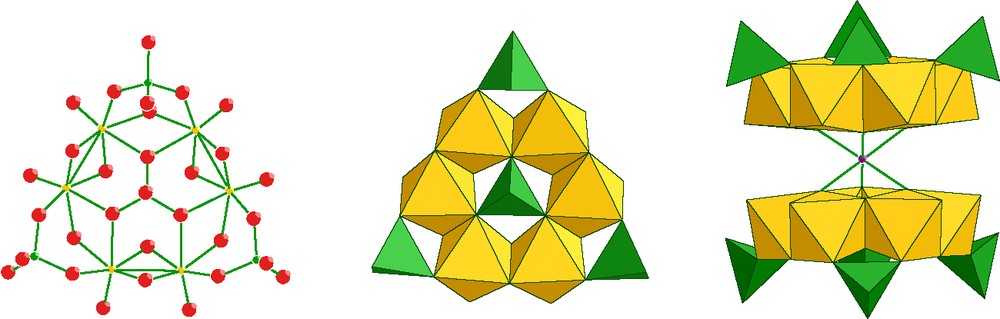
The connections between each building unit are edge sharing. A central phosphato group is encapsulated in the metal ring, the central and the three peripheral phosphates lying in the same side of the six-metal plane. Dimeric sandwiched species were characterized with M2+ cations (M = Cr, Mn, Fe, Co, Ni, Zn, Cd) or with sodium. In the solid state, the transition-metal ions M2+ trapped in the sandwich are isolated from each others that do not confer any specific magnetic properties to the solid. By controlling the initial pH and the stoichiometry of the reaction mixture {Mo2O4}2+/phosphate/Co2+ ions, we succeeded to isolate by hydrothermal synthesis a two-dimensional cobalto-molybdenum phosphate constructed from unprecedented large structural groups [8]. The structure can be described as building units formed of sixteen molybdenum atoms and four tetramers of cobalt forming the 19 Å anionic wheel [(Mo2O4)8(HPO4)14(PO4)10Co16(H2O)20]10–.
The sixteen MoV are arranged in four tetramers linked together by phosphates that form a hexadecamer of MoV. Each Mo-tetramer results from the association of two {Mo2O4} initial building blocks. Two phosphato groups link a tetramer of molybdenum to two tetramers of cobalt. A cobalt-tetramer consists in four distorted edge-sharing octahedra forming a square. The Co2+ ions are bridged by six phosphates and a water molecule, the coordination of the cobalt ions is achieved by a terminal water molecule and an oxygen atom of a {Mo2O4} group.
A striking detail is the presence in the center of the wheel of a floating [Co(H2O)6]2+ octahedron stabilized by hydrogen bondings with the closest phosphato groups. Each wheel is connected to four other wheels by four cobalt dimers through peripheral phosphato groups. The wheel connections ensured by dimeric Co2+ octahedra lead to the layer structure depicted in Fig. 3.
Connecting scheme in [(Mo2O4)8(HPO4)14(PO4)10Co16(H2O)20)]10–, Mo are orange, Co octahedra are violet, and PO4 are green.
The magnetic behavior was studied in the range 2–300 K, and the result is given in Fig. 4 as the plot of XMT versus T (XM is the magnetic susceptibility per unit). The curve continuously decreases upon cooling from 300 K (XMT = 55.54 cm3 mol–1 K, g = 2.32) to 2 K (XMT = 11.75 cm3 mol–1 K).
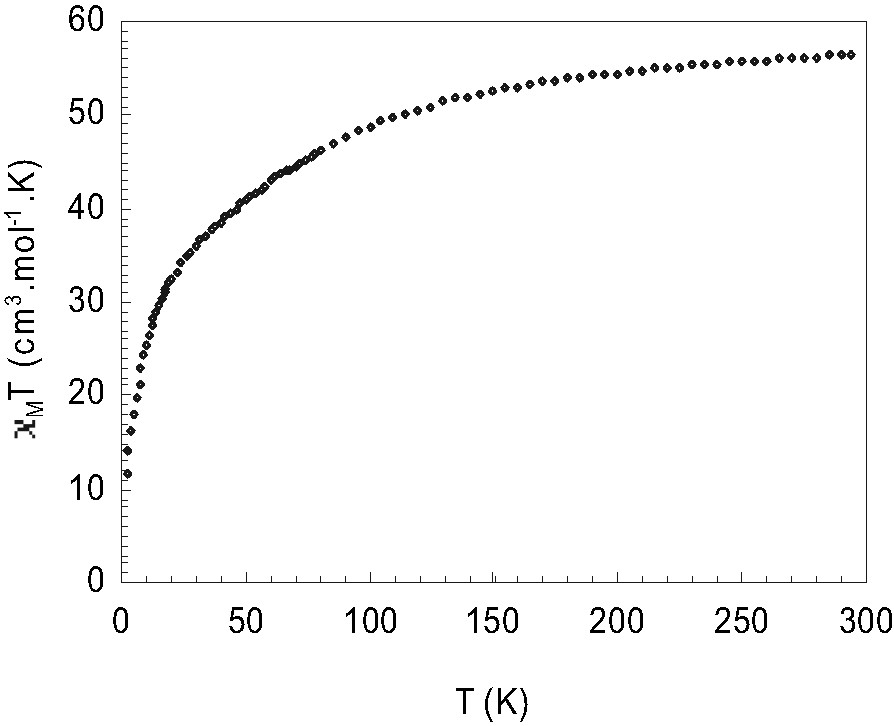
Variation of XMT versus T for [(Mo2O4)8(HPO4)14(PO4)10Co16(H2O)20)]10–.
Because of the MoV–MoV metal–metal bond, the only magnetically active species are the Co2+ ions. The low-temperature XMT is significantly lower than the theoretical value deduced for isolated Co2+ ions for any values of Δ/λ, where λ refers to the spin-orbit coupling parameter and Δ to the ligand-field splitting parameter. This clearly indicates relatively strong antiferromagnetic Co–Co interactions. This result differs from that reported for the tetrameric cobalto-polyoxometalate [Co4O14(H2O)2(PW9O27)2]10– [9], which exhibit ferromagnetic coupling. The difference comes from the geometry of the cobalt-tetramer, here the Co–O–Co angles (94–108°) do not permit the magnetic orbital at the bridges to be non orthogonal.
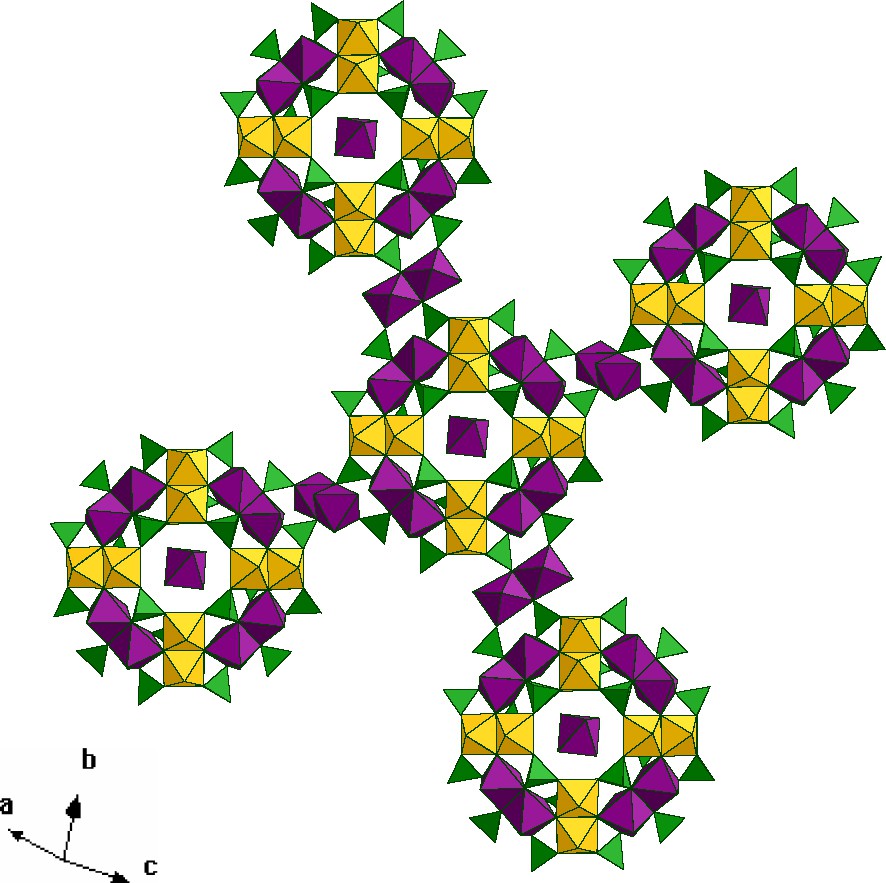
By increasing the pH value of the reaction solution (from 2.0 to 3.9) and decreasing the Co-stoichiometry another layered material was isolated, both similar and different from the former. The structure of the building wheel is strictly analogous, but the wheels are differently interconnected, which leads to a drastic change in the two-dimensional structure (Fig. 5). Each wheel is connected to four other wheels by four single cobalt ions in a distorted tetrahedral environment, which imposes a non-planar arrangement.
View of [(Mo2O4)8(HPO4)14(PO4)10Co16(H2O)20)]10– units connected by tetrahedral {Co4O4} groups, the color scheme is similar to Fig. 3.
With Ni2+ the sixteen-metal building unit is similar, but each wheel is connected to four others by two dimers and by two monomers of nickel, respectively. In fact, the Ni-layered structure exhibit hybrid connections reminiscent of the two former Co-compounds. The magnetic behavior of this material is complex, additional informations are available in the literature [9].
3.2 Polyphosphates as assembling groups in standard conditions
Polyoxometalates containing polyphosphato groups are rare. Kortz and Pope reported in 1994 the first X-ray structural determination of a polyoxomolybdate containing the pyrophosphato (P2O7)4– ligand. Since then, only two other structures have been reported [10].The lack of examples concerning this class of compound and the absence of MoV pyrophosphate complex is justified by the works of Weil–Malherbe and Green [11] who noticed that molybdenum(V) catalyzes the hydrolysis of pyrophosphate into monophosphate in water. This reaction is optimal at pH 2 with a secondary maximum at pH 5.5. We have determined a domain where the hydrolysis of pyrophosphate by MoV is minimum. This led to the first pyrophosphate/MoV complex Na24{Na4(H2O)6⊂[(Mo2O4)10(P2O7)10(CH3COO)8(H2O)4]}·97 H2O, a highly charged and highly hydrated complex formed of two nearly perpendicularly interconnected wheels, an unprecedented topology for an inorganic compound, see Fig. 6. The synthesis was performed in sodium acetate buffer. This is justified considering that the rate of hydrolysis of (P2O7)4– is lowered at the pH imposed by the acetate buffer and that acetate can stabilize the [Mo2VO4(μ-CH3COO)]+ dimer [12] which possesses two free sites in cis-position on each MoV available for the formation of large rings. The largest wheel (noted {Mo12}) contains twelve MoV centers while the smallest (noted {Mo8}) is formed of eight MoV atoms. All Mo-centers are in a distorted octahedral environment with a short terminal Mo = O bond. As usually observed [11,12] the MoV atoms are arranged by pairs, forming the diamagnetic dinuclear unit {MoV2(μ-O)2O2}, with an average Mo–Mo distance of 2.54 Å characteristic of a Mo–Mo bond. These dimers are linked by two types of pyrophosphates. Six pyrophosphates connect two dimers, while four others bridge three dimers ensuring the connection between the two wheels. These two wheels are nearly perpendicular with an angle of 92.8°. The coordination of four of the six dimers of the {Mo12} moiety is achieved by a μ-O2 acetato anion, the coordination of the two remaining being completed by water molecules.
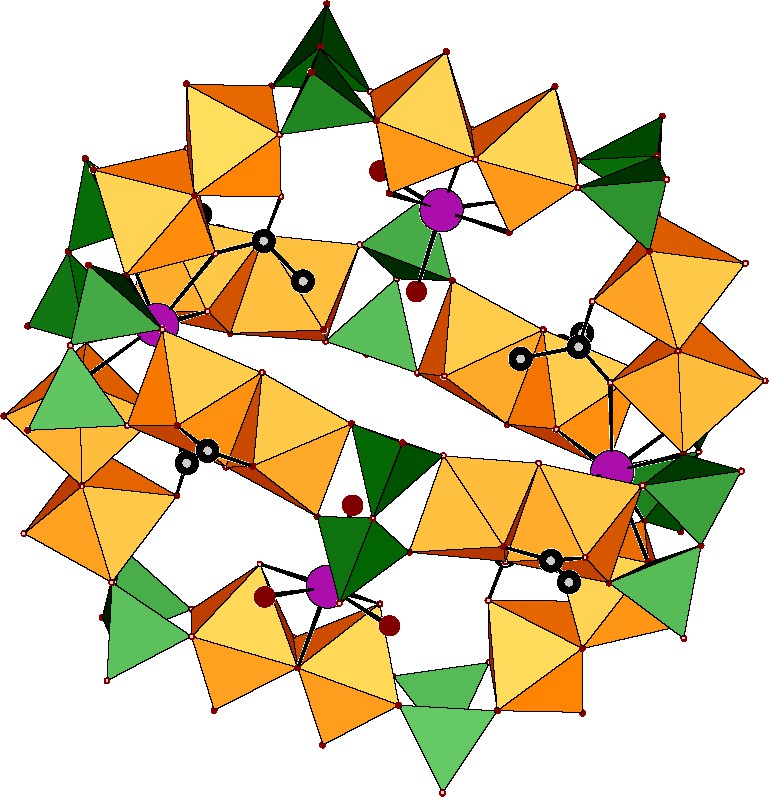
Representation of the double wheel in {Na4(H2O)6⊂[(Mo2O4)10(P2O7)10(CH3COO)8(H2O)4]}24–, same color code.
These exogenous ligands are crucial since they strongly influence the curvature of the and consequently the topology of the whole edifice. This can be emphasized by the distances between the axial ligands in a {MoV2O4(H2O)2} dimer (d(Ot···Ot) = 2.910(9) Å and d(OH2···OH2) = 3.144(9) Å) and in the {MoV2O4(CH3COO)2} dimers (d(Ot···Ot) = 3.172(9)–3.254(10) Å and d(OOCH3···OOCH3) = 2.171(9)–2.207(10) Å). In the {Mo12} wheel, the Mo = O groups are directed toward the outside of the cavity. The situation is quite different for the {Mo8} wheel: each dimer is coordinated to an acetate anion and the Mo = O groups are directed toward the inner cavity. All the pyrophosphato groups are fully deprotonated, which is consistent with the presence of 28 Na+ cations in the lattice. Four of these cations are located inside the cavity and two others at the intersections of the wheels, connecting four {MoV2O4(CH3COO)2} fragments via eight oxygen atoms (d(Na–O) = 2.403(7)–2.516(7) Å). These bonded sodium play a key role in the stability of the host acting as a cryptand anion [13]. Two additional alkaline cations are linked to the water molecules of the {MoV2O4(H2O)2} dimers, the remaining cations being located outside the cage. Finally, a vacant site remains available at the center of the polyanion.
The 31P NMR spectrum of the double wheel in acetate buffer exhibits at room temperature three resonances located at δ1 = 3.03 ppm, δ2 = 1.66 ppm and δ3 = –0.64 ppm with relative intensity of 2:1:2, respectively. This result is in agreement with the solid-state structure. The spectrum remains unchanged over several weeks, showing the high stability in this medium. In pure water, the 31P NMR spectrum presents a complex pattern, indicating decomposition in this medium (Fig. 7).
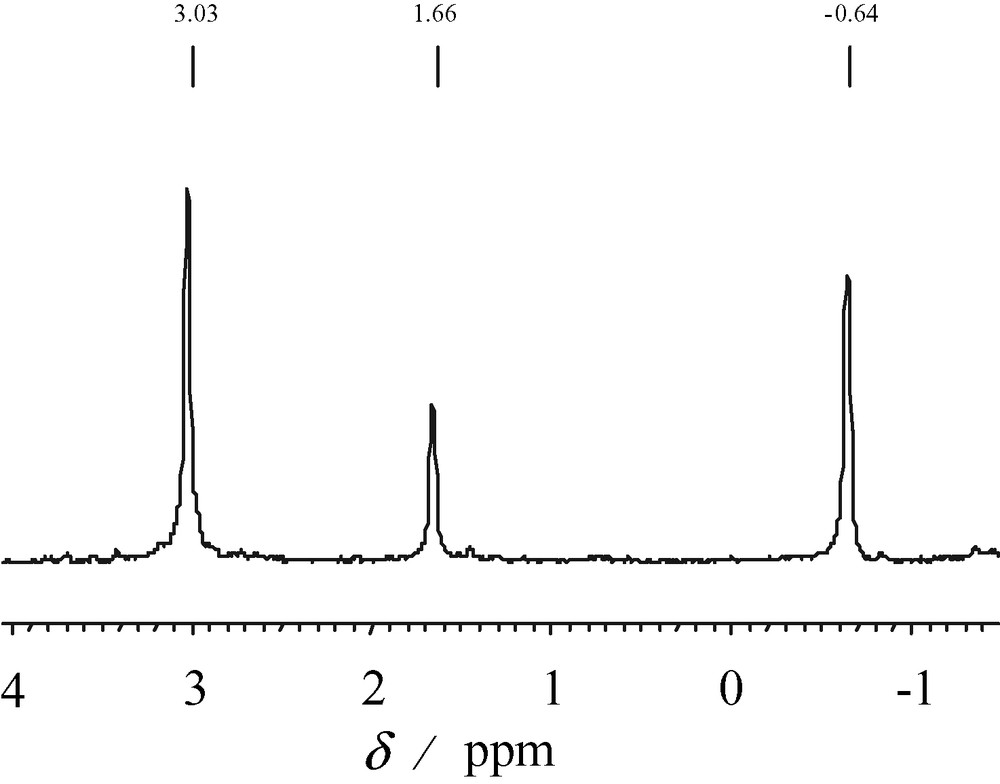
31P NMR spectrum of the double wheel dissolved in a solution of phosphate 0.5 M.
Organophosphonato ligands like [O3PCH2PO3]4– are interesting since they have a geometry quite similar to that of diphosphates, and they present the advantage to be stable toward hydrolysis. Thus, they represent an alternative way to prepare [O3PXPO3]4–/molybdenum systems. To date, only two methylenediphosphonate compounds have been isolated, the fully oxidized [Mo6O18(O3PCH2PO3)(H2O)4]4– anion [14] and the partially reduced [MoV6MoVIO16(O3PCH2PO3)3]8– compound [15]. In both cases, the structure consists in six coplanar molybdenum ions forming a ring.
[Mo2O4]2+ reacts at RT with methylene diphosphonate yielding Na24[Na4(H2O)6{(MO2O4)10(O3PCH2PO3)10(CH3COO)8(H2O)4}].95H2O. This compound [16] is isostructural to the former pyrophosphate double wheel and is stable only in a solution of acetate/acetic acid buffer. In pure water, bridging acetates exchange with water molecules resulting in other different soluble species evidenced by 31P NMR.
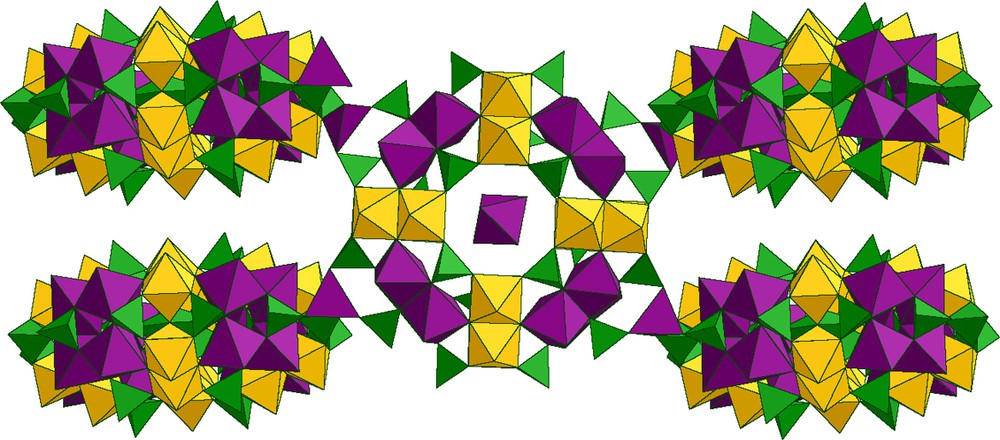
The [Mo2O4]2+ dioxocation has been condensed with [O3PCH2PO3]4– in the presence of templating exogenous ligands such as carbonate or molybdate. Some of the results obtained are given in Fig. 8, as an illustration of the versatility of the chemical system. The common point to all these structures is their cyclic arrangement resulting from the association of Mo(V)–Mo(V) dimers with diphosphonate ligands.
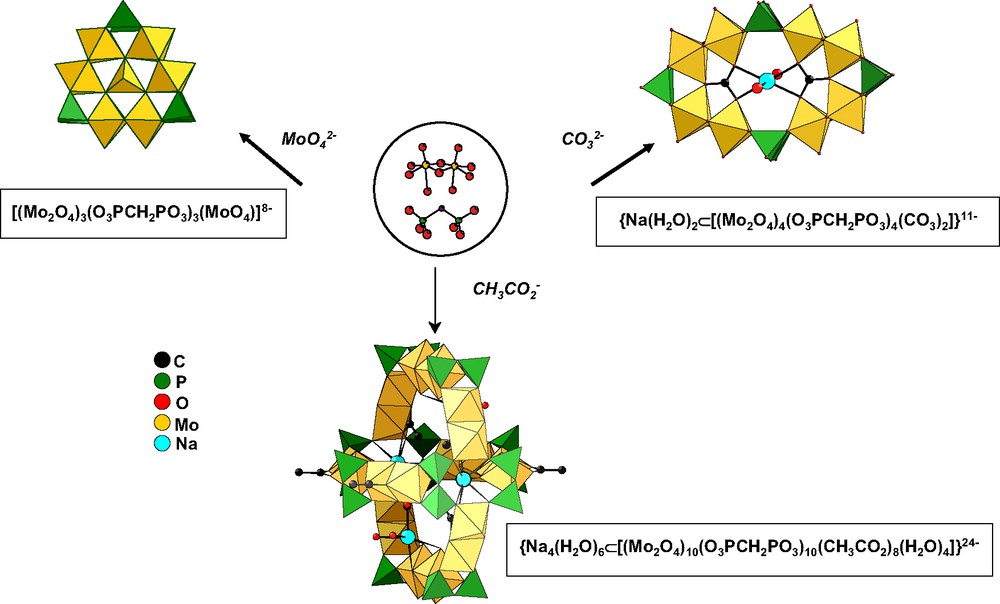
Presentation of the compounds obtained by reaction of methylenediphosphate and [MoO2O2]2+ in the presence of various templating groups.
With molybdate [MoO4]2–, a wheel formed of Mo(V) octahedra, alternately linked by edges and corners, encapsulating a central tetrahedral [Mo(VI)O4} group is formed. The overallstructure is stabilized by three peripheral diphosphonates (Fig. 8).
Carbonates led to a wheel containing four Mo(V) dimers and four [OPCH2PO3]2– ligands encapsulating two carbonate ions and a central {Na(H2O)2}+ group (Fig. 8).
4 [Mo2O2S2]2+ based wheels
4.1 Halide recognition
The presence of a double sulfido bridge confers to the [M2O2S2]2+ building block specific coordination properties quite different from that reported for the oxo analogue [Mo2O4]2+. The cyclic architectures described below exhibit supramolecular properties potentially relevant to anion recognition, anion-templated synthesis and 3-D structures [17].
A yellow powder was precipitated by direct addition of potassium hydroxide to a mixture of the dithiocation and potassium iodide about pH 2–3. After crystallization in pure water, the neutral dodecameric wheel, [Mo12O12S12(OH)12(H2O)6], noted {Mo12}, was isolated [18]. The structure is that of a Mo12-ring formed of six {Mo2O2S2} fragments, linearly connected by double hydroxo-bridges (Fig. 9).
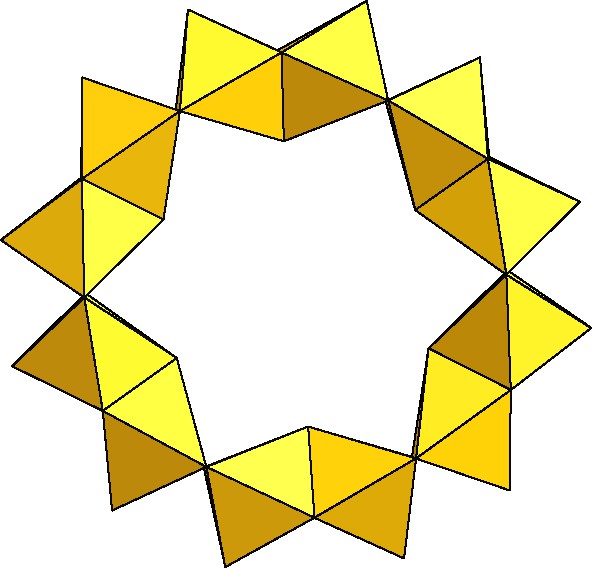
Polyhedra representation of the neutral [Mo12S12O12(OH)12(H2O)6] wheel.
The cyclic backbone {Mo12O12S12(OH)12} is neutral and delimits a central open cavity of about 11 Å, lined by six water molecules. The inner water molecules are labile enough to be exchanged for specific anions, suggesting the cavity exhibits a cationic character, induced by the presence of twelve Mo(V). This situation is quite different with that encountered with crown ether compounds where the cavity is lined by donor oxygen atoms giving the cavity an anionic character. Only the four equatorial aquo ligands of the initial dithiocation are involved in the olation reaction. The four equatorial aquo ligands are ionized below pH 2, provoking the oligomerization process, while the two axial water molecules are unaffected in the Mo12-ring. The crystallization of {Mo12} in DMF containing tetrabutylammonium iodide or tetraethylammonium chloride leads to Cat2[X2Mo10S10O10(OH)10(H2O)5] with Cat = Nbu4 and X = I, or Cat = Net4 and X = Br, respectively [19], see Fig. 10. The two molecular structures are similarly built on a neutral decanuclear ring exhibiting the same connection scheme to the previous Mo12-ring.
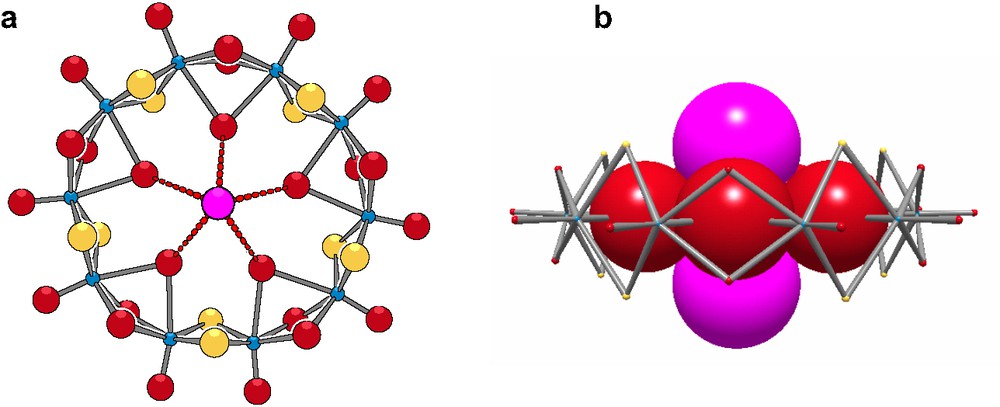
Representations of [Cl2Mo10S10O10(OH)10(H2O)5]2–; (a) in ball and sticks; (b) space-filling showing how the iodide is deeply engaged in the ring.
In the solid state, two halide ions are symmetrically located on both sides of the mean plane defined by the 10 Mo atoms. The distances between the two halides and the five-oxygen atom of the ring are short enough to suggest that the stability of the supramolecular arrangement is ensured by an H–bonding network involving the protons of the inner water molecule.
In water, successive equilibria based on halide exchanges permit to obtain the iodide free neutral {Mo12} cluster. In contrast, the presence of halide ions (Cl– or I–) and/or aprotic solvent are required to maintain the {Mo10} backbone. Potentiometric titrations of the {Mo10} wheel in the presence of halides show successive ionizations of the five inner water molecules. The five related acidity constants range from 4 to 9. On this occasion, within pH 6–6.5, the [ClMo10S10O10(OH)12(H2O)3]3– anion was characterized. The molecular structure resembles that of the parent, but on replacing two inner aquo ligands by two hydroxo groups, the inorganic cycle interacts with only a single chloride. Halide ions can be considered as templates for imposing the nuclearity of the wheel, which can swing from 10 to 12. Thus, the ability of the cyclic rings to self-rearrange supported by the possibility of such systems to bind soft bases such as halides, open some perspectives in the field of host–guest chemistry.
4.2 Caboxylates as template
The previous results open the way to a cycle-based chemistry, the purpose being to synthesize à la demande rings of different shapes and sizes. The possibility to tune the size and shape of the ring via an adapted template is an exciting challenge. Owing to the reactivity of the cavity of the ring, the use of linear dicarboxylates, oxalate (C2O42–), glutarate (H6C5O42–) and pimelate (H10C7O42–) ions proved to be a good tool to illustrate the influence of the length of the alkyl chain on the size and shape of the wheel. Reaction of the dithiocation with oxalate, glutarate and pimelate ions, led to [Mo8S8O8(OH)8(C2O4)]2– noted [Mo8–ox]2–, [Mo10S10O10(OH)10(H6C5O4)]2– ([Mo10–glu]2–), and [Mo12S12O12(OH)12(H10C7O4)]2– ([Mo12–pim]2–) anions, respectively [20]. This series of compounds [Mo8–ox]2–, [Mo10–glu]2– and [Mo12–pim]2–, nicely illustrates the possibility to vary the size of the ring in a very simple way. The molecular architectures given in Fig. 11, consist in a neutral inorganic ring {Mo2nO2nS2n(OH)2n}, n = 4, 5 or 6 for ox2–, glu2– or pim2–, respectively, encapsulating the different guests.
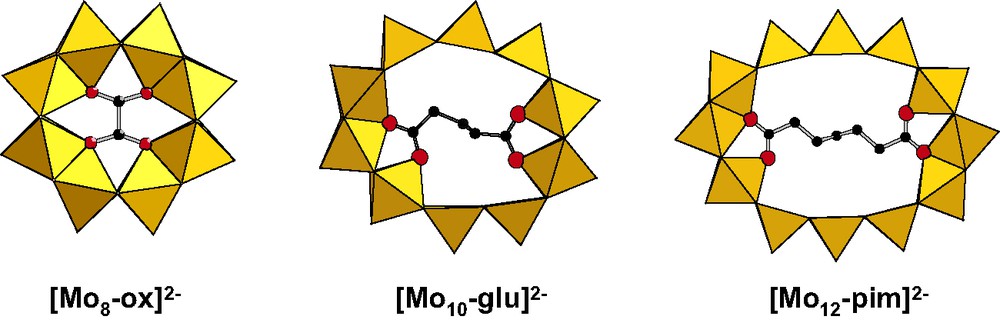
Influence of the size of the carboxylate guest on the size and shape of the {Mo2nS2nO2n(OH)2n(H2O)n} cyclic backbone, ox = oxalate, glu = glutarate, pim = pimelate.
The cyclic skeleton of [Mo8–ox]2– contains eight octahedral molybdenum atoms, each oxygen atom of the two equivalent carboxylate groups is doubly bonded to two adjacent Mo atoms. The structures of [Mo10–glu]2– and [Mo12–pim]2– are distorted but can be considered as formally deriving from those of their perfectly circular Mo10 and Mo12 parents. The encapsulated dicarboxylate groups diametrically coordinate two Mo atoms. No inner water is present in the cavity of [Mo10–glu]2– and [Mo12–pim]2–. This illustrates the versatility of molybdenum, which can adopt either an octahedral, either a square pyramidal coordination. The possibility for MoV to be sixfold or fivefold coordinated gives a great flexibility to these architectures illustrated by the values of the Mo–Mo–Mo angles which can vary, in the same structure, from 135° to 180°. The weaker values are observed at the diametrically opposite carboxylate groups. The organic carboxylate acts as a pincer which distorts the ring. The cycle adapts its geometry to that of the encapsulated molecule. Aqueous solution 1H NMR studies confirmed the presence of the encapsulated organic chain, while electrospray mass spectrometry investigations confirm the crystal structures are maintained in solution.
With trimesate (1,3,5 methylcarboxylato benzene), the perfectly circular dodecameric ring Mo12 is obtained relatively to the symmetrical shape of the trimesate. The encapsulated trimesate lies in the mean plane defined by the twelve molybdenum atoms. The ring ideally fits with the shape of the guest giving the circular wheel represented in Fig. 12.
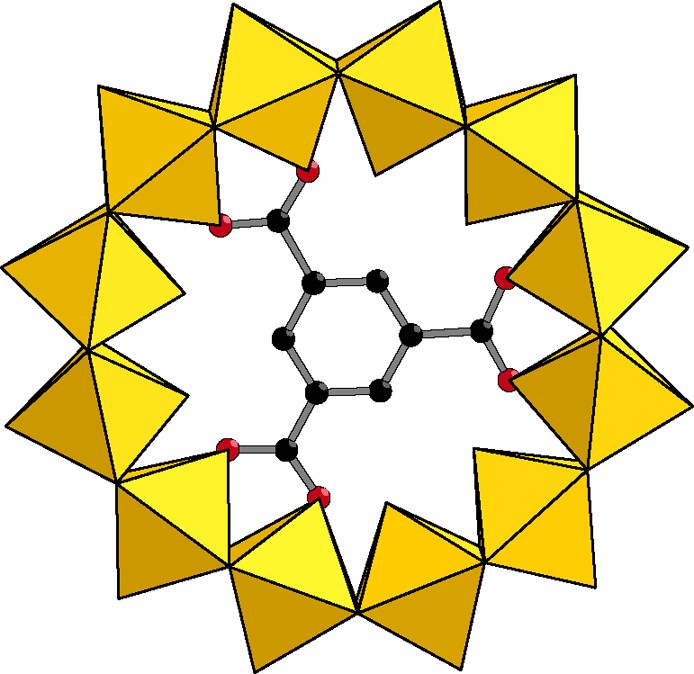
Trimesate, a perfect cyclic directing template.
Now, we are convinced that the nuclearity of the cycle can be tuned via the control of the steric strains imposed by the shape and size of the template. An additional and critical parameter is the number of templates, which can be introduced within the ring.
4.3 Phosphates as template
Phosphates are interesting species for their chelating and acidobasic properties, small size and tetrahedral shape. [(HPO4)Mo10S10O10(OH)10(H2O)3]2– has been prepared [21] in nearly stoichiometric conditions in phosphate from dilute aqueous solutions. The structure is that of a neutral decanuclear ring {Mo10O10S10(OH)10} encapsulating a single hydrogeno-phosphate [HPO4]2– anion. [(HPO4)2Mo12S12O12(OH)12(H2O)2]4– was obtained from more phosphate concentrated solutions [21]. The twelve metal ring encapsulates two hydrogeno-phosphato groups [HPO4]2–. The chelating effect resulting from the presence of two phosphate groups strongly distorts the ring from circular to elliptical as represented in Fig. 13.
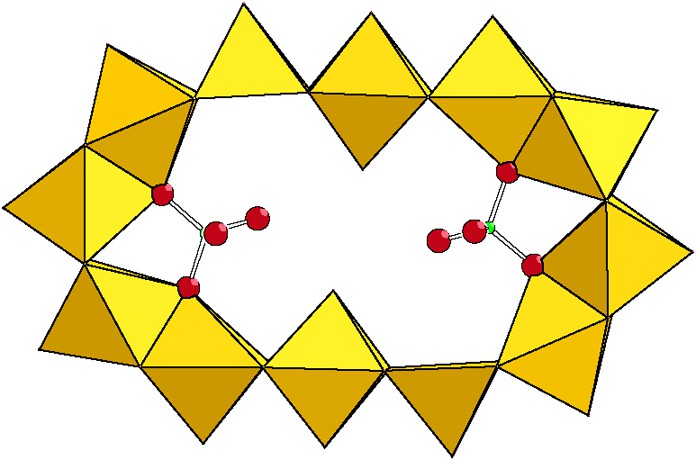
Elliptical shape of the {Mo12S12O12(OH)12(H2O)10} backbone after insertion of two phosphates.
The flattening of the ring shape is attributed to electrostatic repulsions occurring between the diametrically opposed phosphate groups and to their coordination pincer effects. Internal hydrogen bonds between the deprotonated P = O bonds and terminal inner water molecules bring additional stability to the elliptical shape. Solutions containing [PMo10]2– and [P2Mo12]4– have been thermodynamically studied by 31P NMR at various temperatures, revealing fast dynamical phosphate exchanges accompanied with the rapid change of geometry between [PMo10]2– and [P2Mo12]4– [21].
A striking feature was discovered by variable temperature 31P NMR and phosphate–arsenate exchange experiments: the phosphate groups rotate in the cavity, hopping on the vacant adjacent coordination sites. In addition, fast dynamical exchanges take place involving uncoordinated and coordinated phosphate ions in the Mo10 and Mo12 wheels. The phosphate-containing rings system confirms our first hypotheses about the relationship between the ring size and the number of encapsulated template-agent: a single phosphate stabilizes a ten-metal ring, while the insertion of the second one provokes the ring expansion to twelve metals.
In more concentrated phosphate solutions, the hexanuclear [(HPO4)4Mo6S6O6(OH)3]5– anion is exclusively formed in solution (31P NMR) and characterized in the solid-state by X-ray diffraction methods [22]. The overall geometry of the sulfido-{P4Mo6} anion is quite similar to that of the oxo-{P4Mo6} represented in Fig. 2, but its reactivity is drastically different. A central phosphate is encapsulated in a six-metal ring that yields a quite close compact arrangement of oxo and sulfido ions. The arsenate analogue [(HAsO4)4Mo6S6O6(OH)3]5– was also structurally characterized.
The three peripheral phosphates are labile; they can be easily replaced by other functional groups. The mechanism of the substitution reaction was established by 31P NMR spectroscopy by replacing phosphate for acetate groups. The exchange steps are successive with formation of mixed acetate-phosphate species. The quantitative treatments of the data pointed out that the relative affinity of the two substituting groups is essentially related to their own acidity constants. In Fig. 14 the resulting diagram of speciation is given.
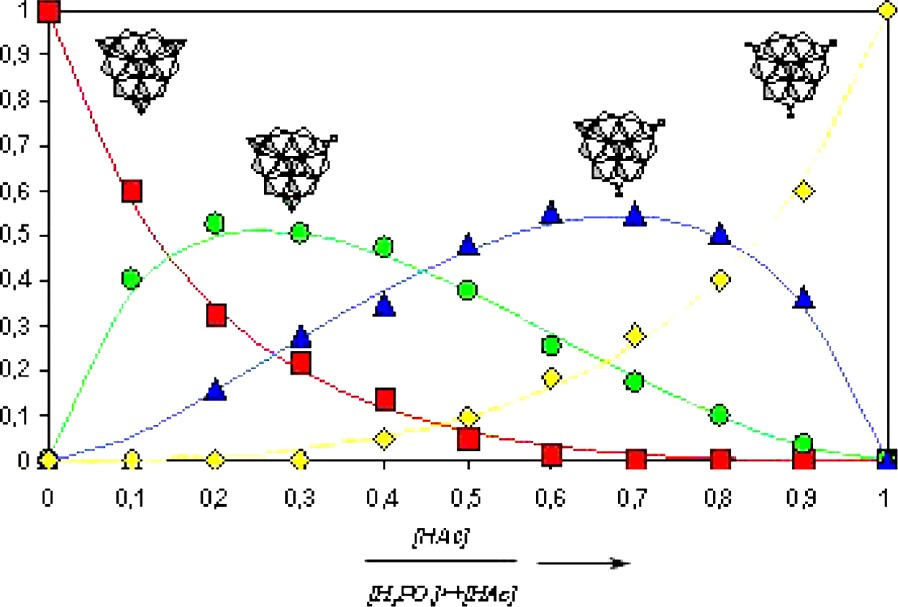
Speciation diagram of the HPO42–/CH3COO– exchange in solution for [(HPO4)4MoMo6S6O6(OH)3]5–.
The selective replacement of sulfur atoms within the {Mo2S2O2} fragment by oxygen atoms was obtained by controlled hydrolysis. The reaction takes place in drastic conditions (80–100 °C) and in agreement with 31P NMR results, the nucleophilic substitution revealed to be regioselective. In a first step, only the three coplanar sulfur atoms located at the opposite side of the four phosphate groups are concerned by the substitution. The other three sulfur atoms are protected by the four bulky phosphates located nearby. The mechanism of substitution was confirmed by the resolution of the structure of the half-desulfurated [(HPO4)4Mo6S3O9(OH)3]5– anion, which supports the previous 31P NMR observations in solution.
4.4 Metalates as template
At this step, we know that organic and p-block oxo-anions can serve as ring templating groups. In fact, more sophisticated templates, like transition-metal derivatives have been also used as structuring guests. With [MO4]2– (M = Mo or W) tetra-oxometalate ions, the self-condensation of the dithiocation [Mo2O2S2]2+ gives the mixed metal [Mo8S8O8(OH)8(HMO5(H2O))]3– compounds [23] containing a central {MVIO6} octahedron encapsulated in an octanuclear ring {Mo8S8O8(OH)8}. This arrangement resembles that previously described for oxalate ion, see § 4.2. The geometry of the encapsulated Mo(VI)-moiety has changed from tetrahedral to octahedral. Although the anion consists in a mixed valence MoVI/MoV system, the electronic spectrum does not exhibit any intervalence charge transfer (IVCT) from peripheral MoV atoms toward the central MoVI. The eight Mo(V) electrons are firmly trapped as pairs in the metal–metal bonds, and in addition the junctions between the MoV and MoVI centers are ensured through long Mo–O bond (2.2–2.4 Å) in trans position to the Mo=O double bond (trans effect). Thus, such compounds exhibit strongly trapped mixed valence for ranging in the class I of Robin and Day. The tungsten analogue was obtained and characterized.
A family of octanuclear wheels has been obtained by crystallization of [Mo8S8O8(OH)8(HMO5(H2O))]3– and [Mo8S8O8(OH)8(C2O4)]2– in an aqueous solution of LiCl, NaCl, KCl, and RbCl. Single crystals X-ray diffraction studies on these solids showed that the alkali salts exhibit a similar 3-D structure represented in Fig. 15.
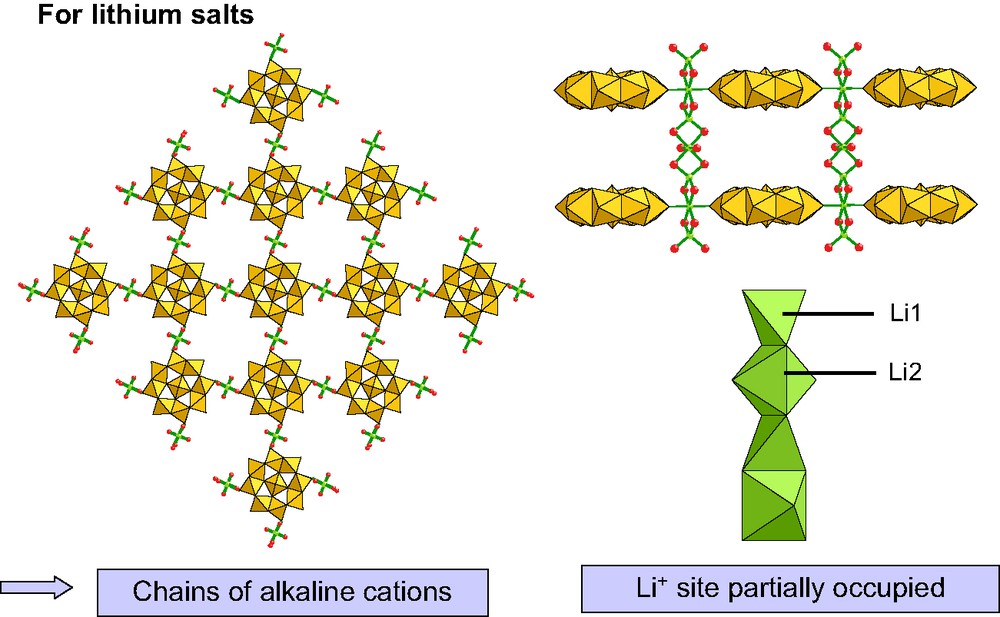
View of the alkaline pillars decorated by [Mo8S8O8(OH)8(C2O4)]2– anionic wheels.
Alkali cations are located in columns to which the anionic wheels are anchored [24]. Ionic conductivity measurements on pressed pellets revealed two different behaviors. The lithium salts of {HMo8M3}– (M = Mo, W) are moderately good proton conductors at room temperature, σ = 10–5 S cm–1. The profile of conductivity as a function of relative water pressure showed that the conductivity is related to surface motion.
In contrast, the lithium salt of {Mo8ox}2– competes with the best solid lithium conductors at room temperature, σ = 10–3 S cm–1. The mobility of lithium cations along 1D channels was confirmed by 7Li NMR experiments [24].
5 Extended frameworks
We limit our description to only one example [19]. The supramolecular arrangement of the mono chloride complex [ClMo10S10O10(OH)12(H2O)3]3– described in Section 4.1 was crystallized as potassium salt. K+ polyhedra form planes parallel to (010) that delimit 10 potassium ring domains, filled by a first set of wheels (Fig. 16). These planes are connected through a second set of wheels leading to a remarkable 3-D array.
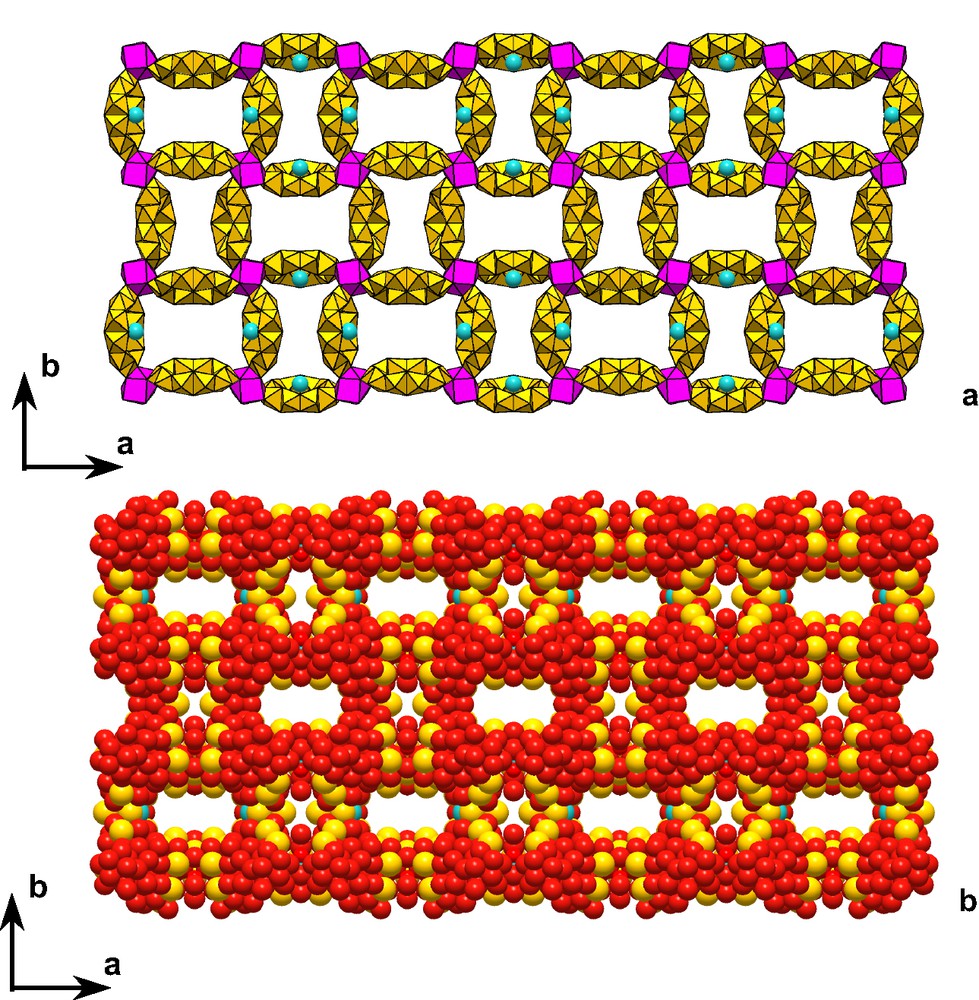
View of the 3D-arrangement of [ClMo10S10O10(OH)12(H2O)3]3– in the solid state. Channels contain only water molecules. (a) Polyhedral representation; (b) space-filling representation.
The resulting grid delimits large ring-channels with wide voids (16.4 × 8.0 Å), filled by water, labile enough to be exchanged. Studies are in progress to investigate the thermal stability, water removal and BET surface of the array.
6 Conclusion
Owing to the fact that the [M2O2E2]2+ cations (E = O, S) were evidenced in numerous complexes, nobody could imagine that these fragments could serve as precursors for condensation, offering to the chemist a versatile and powerful tool for the edification of series of original compounds. Here, the successful strategy we described can be stated as a building block approach. Thus, cyclic oxo-thio-metalates represent an important new class of compounds, that can be ranked in the wide field of the early transition metal block named polyoxometalates. The first bases of their chemistry are laid. The syntheses are facile, the precursor can be obtained on a one fifty gram scale and products are obtained by simple admixture of the precursor and the template in water. In most cases the yield is quantitative. In fact, [M2O2E2]2+ E = O, S revealed to be a magic cation.