

1 Introduction
Difluoro-boradiaza-s-indacenes, commonly named boron–dipyrromethene dyes (Bodipy) [1], are widely employed as useful fluorescence probes for biomolecules labeling [2], and for sensing of calcium ion [3], nitric oxides [4], protons [5] or various cations [6,7] by opto-electronic switching. The attractive features of these dyes stem from the facile modification of their structures, providing opportunities to finely tune their properties and to incorporate recognition sites for a variety of analytes. These dyes combine sharp absorption bands (fwhm ≈ 25–35 nm), high molar absorption coefficients (ɛ = 40,000–110,000 M−1 cm−1), large fluorescence quantum yields (Φ = 60–90%), and relatively long excited-state lifetimes (1–10 ns). These dyes exhibit excellent chemical and photochemical stability in solution and in the solid state, and for some of them valuable charge-transfer properties. Furthermore, their optical properties are sensitive to modifications around the pyrrole core [8], the central meso position [9,10] and the boron atom [11]. In addition, the dyes have good solubility in organic solvents and are thus amenable toward chromatography on silica or alumina. The high purity and neutrality of these interesting fluorophores allow vacuum deposition in conventional equipment. Extensive efforts have therefore been devoted to the design and preparation of sophisticated dyes for the use as chromogenic probes [12], fluorescent switches [13], electro-chemiluminescent materials [14,15], laser dyes [16], sensitizers for solar cells [17], fluorescent labels for biomolecules [11], drug delivery agents [18], and as electron-transfer probes for radical ion pairs generated by local electric fields [19].
In some exceptional cases, Bodipy has been used as an antenna transferring its excitonic energy to acceptor frameworks such as zinc porphyrins [12,20–22] or perylene dyes [23]. Extensions of these pioneering studies have led to the development of systems capable of absorbing light and transferring the excitation energy to an acceptor able to promote photo-induced electron-transfer processes over long distances, leading to long-range charge separation mimicking the natural photosynthetic reaction centre [24,25]. The propensity of Bodipy to transfer an electron under irradiation has been extensively exploited in chemosensor systems [26]. Electroluminescence from borodipyrromethene dyes doped into a polyvinyl–carbazole host polymer [27] or thin films of aluminium-complexes has previously been observed in OLEDs [15]. Electrogenerated luminescence has also been investigated in acetonitrile solution by pulsing the working electrode between the first oxidation and reduction peaks of the Bodipy [14]. The well-defined molecular structure of Bodipy makes it relatively easy to establish firm structure–reactivity relationships. Recently, novel Bodipy architectures enabled their incorporation into supramolecular assemblies, such as liquid-crystalline materials or organo-gelators [28–30], and luminescent phenol–pyridyl–boron complexes displaying bright luminescence have been exploited in white and blue electroluminescent devices [31].
Herein we report new Bodipy derivatives bearing gallate-substituted residues with methoxy groups or aliphatic chains (8, 10, 12, 16 and 20 carbons) well characterized by X-ray diffraction on single crystals, NMR, absorption and fluorescence spectroscopies, and electrochemistry. The methoxy-substituted derivatives gives electroluminescent films either as pure material by vacuum sublimation or by solution processing in PVK polymers.
2 Results and discussion
2.1 Synthesis
The one-pot synthesis of the starting nitro- and dinitrophenyl derivatives of 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene is depicted in Scheme 1. First, the condensation of 2,4-dimethyl-3-ethylpyrrole (also called kryptopyrrole) with the corresponding mononitro or dinitro acyl chlorides is performed in dichloromethane at room temperature [32]. The resulting dipyrromethene salts is then complexed to a BF2 fragment after deprotonation with triethylamine. The desired nitrophenyl-Bodipy cores are obtained in fair yields (55% for the mononitro and 43% for the dinitro compounds). By means of a catalytic hydrogenation over Pd on charcoal, these nitro derivatives of 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene can be easily reduced into the corresponding amines. Interestingly, the use of quite inexpensive reagents, the fair to good yield of each step involved in the synthesis and finally the quite smooth procedure of catalytic hydrogenation, compared to the techniques used to perform hydrogenation on similar compounds [33], render the synthesis of nitro and amino 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene derivatives very accessible.
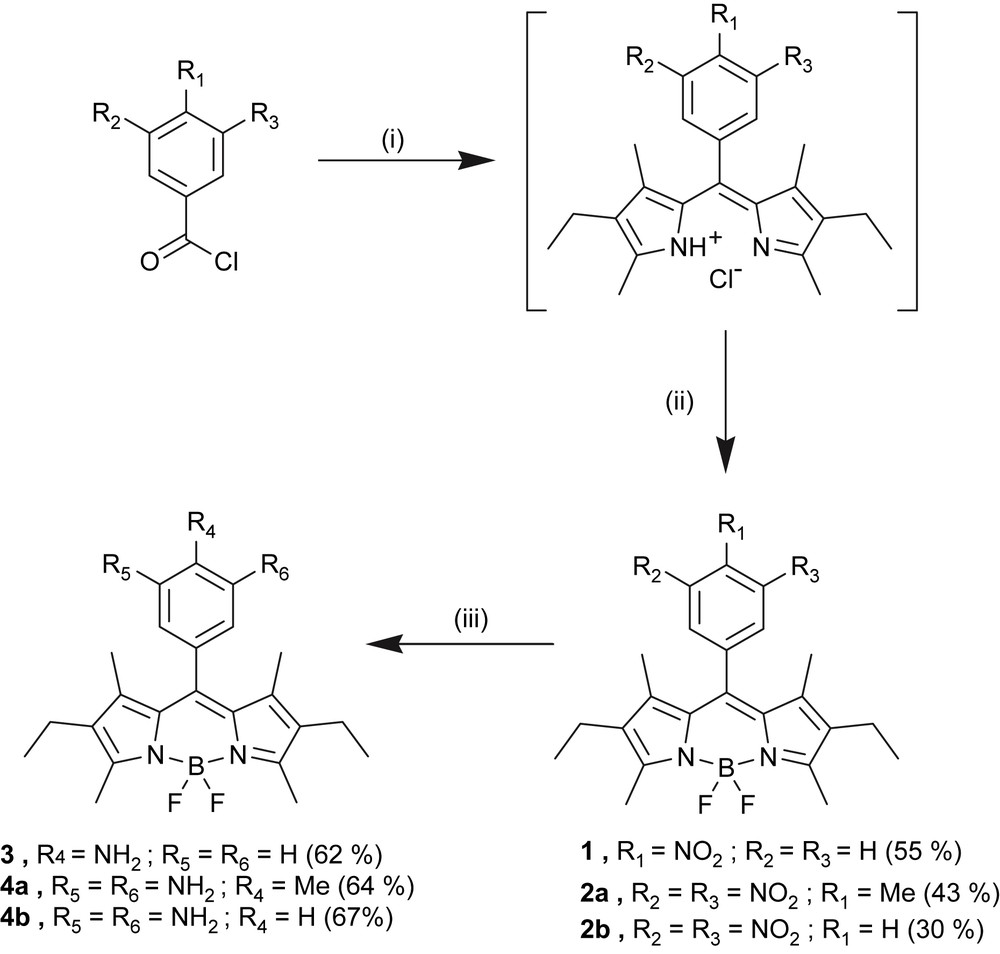
(i) 2,4-Dimethyl-3-ethylpyrrole (2 equiv), CH2Cl2, rt, 3 days; (ii) BF3·Et2O (8 equiv), TEA (6 equiv), rt, 1 day; (iii) H2 (1 atm), 5% Pd/C (0.1 equiv), EtOH/CH2Cl2, rt, 1 day.
Reaction of these amino 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene derivatives with a series of 3,4,5-tris-alkyloxy-benzoic acids leads to the synthesis of a wide range of amido Bodipy derivatives functionalized with straight or branched paraffin chains. Introduction of long carbon chains improves the solubility in common organic solvents, facilitates the evaporation under high vacuum, and results in well-organized solid-state phases by microsegregation between the polar Bodipy core and the apolar chains. The amide bond connecting the fluorophore to the gallate platform was especially introduced to control and stabilize the molecular organization through hydrogen bonding (vide infra). Its formation results from reacting the amino-Bodipy compounds 3 or 4a either with the 3,4,5-tris-alkyloxy-benzoic acid chlorides in presence of triethylamine (TEA) or directly with the corresponding acids in the presence of the acid chloride salt of 1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide (EDC·HCl) and dimethylaminopyridine (DMAP) (Scheme 2). For the phytol strands, a racemic mixture of 3,4,5-tris-(3,7,11,15-tetramethyl-hexadecyl) was used, whereas for citronellol strands the pure (S)-(+)-citronellyl derivative was used [34]. All the compounds were fully characterized by means of NMR and FT-IR spectroscopies, ESI-MS or FAB-MS and elemental analysis.

(i) 3,4,5-Tris-alkyloxy-benzoic acid chloride (1 equiv), TEA, CH2Cl2, rt; (ii) 3,4,5-tris-alkyloxy-benzoic acid (2.2 equiv), EDC·HCl (2 equiv), DMAP (2 equiv), CH2Cl2, rt.
2.2 Optical and electrochemical properties in solution
Spectroscopic data for all compounds are gathered in Table 1. All the compounds show similar absorption patterns which are characteristic of Bodipy fluorophores, and a representative example is depicted in Fig. 1. The absorption spectrum is composed of a strong S0 → S1 (π–π∗) transition located around 525 nm, with molar extinction coefficients ranging from 70,000 to 100,000 M−1 cm−1, in keeping with classical F-Bodipy derivatives [35]. A second absorption band centred around 370 nm is assigned to the S0 → S2 transition of the Bodipy subunit [36]. The third absorption around 275 nm is likely due to π–π* and n–π* transitions localized on the phenyl and dipyrromethene fragments [37]. The amido compounds have high fluorescence quantum yields ranging from 58 to 91%. The weak Stokes' shifts (about 500 cm−1) observed over the whole series of fluorophores is in good agreement with a singlet emitting state. Excitation spectra perfectly match absorption spectra (Fig. 1 as a typical example), which is in keeping with a unique excited state, excluding the existence of a CT transition, despite the presence of both electron-donating and electron-attracting fragments on these fluorophores. Moreover, no quenching of the luminescence by molecular oxygen is observed, excluding the presence of a possible triplet emitting state. The fluorescence decay profiles of these molecules can be fitted by a single-exponential, with fluorescence lifetimes ranging from 2.5 to 9.3 ns (Table 1 and inset in Fig. 1), in line with a singlet emissive state. All fluorescence spectra exhibit nice mirror symmetry with the lowest energy absorption band, meaning that the corresponding transitions involve the same excited state. The absence of fluorescence observed for the nitro compounds could easily be explained within the framework of a photo-induced electron transfer from the Bodipy excited state (a very good reductant about −1.40 V versus SCE) toward the nitro fragments (quite easy to reduce about −1.10 V versus SCE). An estimate of the driving force for PET could be determined by an interplay between optical properties and redox properties and lie within −480 and −650 mV. Finally, most of the radiative rate constants lie within the 1.0–1.2 × 10−8 s−1 range (Table 1). However, the non-radiative rate constants are significantly weaker than the radiative ones, mostly because of the strong quantum yields and little deactivation by side pathways. At this stage of our investigation, we do not have evidence for the formation of an energetically localized low lying excited state or triplet excited state that could quench the fluorescence.
Optical and electrochemical data measured in dichloromethane solution at 298 K
| Compounds | λabs (nm) | ɛ (M−1 cm−1) | λF (nm) | ΦFa | τF (ns) | krb (108 s−1) | knrb (108 s−1) | E0oxy, V (ΔE, mV)c | E0red, V (ΔE, mV)c |
| 1 | 533 | 70,000 | 540 | 0.02 | <1 | – | – | +1.05 (60) | −1.09 (110) |
| −1.33 (60) | |||||||||
| 2a | 535 | 90,000 | – | – | – | – | – | +1.10 (70) | −0.99 (80) |
| −1.32 (88) | |||||||||
| −1.70 (irr.) | |||||||||
| 2b | 536 | 67,000 | 541 | 0.01 | <1 | – | – | +1.13 (60) | −0.85 (90) |
| −1.27 (90) | |||||||||
| −1.49 (90) | |||||||||
| 3 | 526 | 71,000 | 536 | 0.18 | 9.3 | 0.19 | 0.88 | +0.95 (60) | −1.44 (80) |
| 4a | 523 | 90,000 | 538 | 0.01 | 2.5 | 0.04 | 3.96 | +0.95 (irrv.) | −1.40 (70) |
| 4b | 524 | 70,000 | 539 | 0.20 | 1.1 | 0.09 | 9.00 | +0.98 (irrv.) | −1.42 (70) |
| 5 | 525 | 60,000 | 540 | 0.65 | 6.2 | 1.04 | 0.56 | +0.97 (60) | −1.33 (70) |
| +1.54 (irrv.) | |||||||||
| 6 | 525 | 75,000 | 540 | 0.58 | 5.4 | 1.07 | 0.77 | +0.97 (60) | −1.35 (70) |
| +1.51 (irrv.) | |||||||||
| 7 | 525 | 70,000 | 540 | 0.75 | 5.8 | 1.29 | 0.43 | +0.97 (60) | −1.34 (70) |
| +1.51 (irrv.) | |||||||||
| 8 | 525 | 77,000 | 540 | 0.66 | 5.1 | 1.29 | 0.66 | +0.97 (70) | −1.33 (60) |
| +1.52 (irrv.) | |||||||||
| 9 | 525 | 95,000 | 540 | 0.67 | 4.9 | 1.37 | 0.67 | +0.97 (60) | −1.34 (70) |
| +1.54 (irrv.) | |||||||||
| 10 | 525 | 100,000 | 541 | 0.68 | 5.9 | 1.15 | 0.54 | +0.99 (60) | −1.29 (70) |
| +1.52 (irrv.) | |||||||||
| 11 | 526 | 70,000 | 542 | 0.90 | 7.2 | 1.25 | 0.14 | +0.95 (60) | −1.31 (70) |
| +1.54 (irrv.) | |||||||||
| 12 | 526 | 100,000 | 542 | 0.81 | 6.2 | 1.31 | 0.31 | +0.95 (60) | −1.33 (80) |
| +1.52 (irrv.) | |||||||||
| 13 | 526 | 100,000 | 542 | 0.88 | 7.1 | 1.24 | 0.17 | +0.99 (60) | −1.29 (70) |
| +1.53 (irrv.) |
a Determined in dichloromethane solution (c = 5 × 10−7 M) using Rhodamine 6G as reference (ΦF = 0.78 in water, λexc = 488 nm) [38]. All ΦF are corrected for changes in refractive index.
b Calculated using the following equations: kr = ΦF/τF, knr = (1 − ΦF)/τF, assuming that the emitting state is produced with unit quantum efficiency.
c Potentials determined by cyclic voltammetry in deoxygenated CH2Cl2 solutions, containing 0.1 M TBAPF6, at a solute concentration of 10−3 M, at 20 °C. Potentials were standardized using ferrocene (Fc) as internal reference and converted to SCE assuming that E1/2 (Fc/Fc+) = +0.38 V (ΔEp = 70 mV) vs SCE. Error in half-wave potentials is ±10 mV. Scan rate 200 mV/s. When the redox process is irreversible the peak potential (Ecp) is quoted. All reversible processes are monoelectronic.
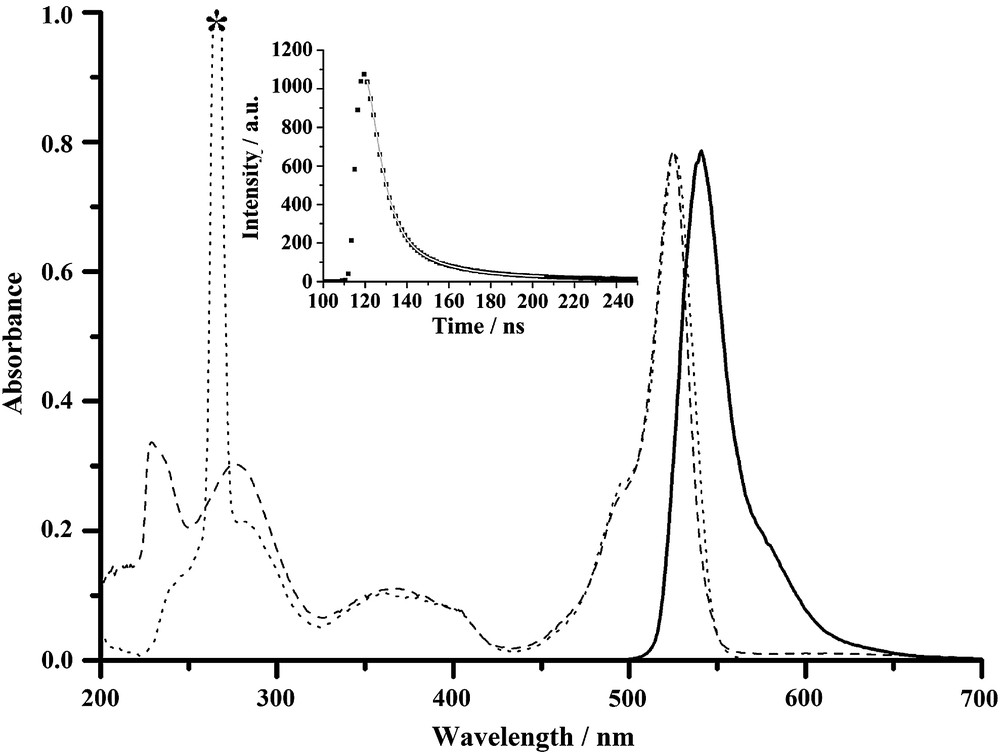
Absorption spectra for 5 (absorption in dashed line, excitation in dotted line), and emission spectra in solid line (λexc = 490 nm). All spectra were measured in CH2Cl2 at rt (c = 5 × 10−7 M). Inset: measured intensity decay of 5 at 540 nm (λexc = 490 nm) and its corresponding monoexponential fit (solid line). * Excitation harmonic.
2.3 Electrochemical properties
The electrochemical properties were determined by cyclic voltammetry in dichloromethane solution. Table 1 lists the potentials (relative to the SCE reference electrode) for the waves that were observed in the +1.6 to −2.0 V window. First, for all of the compounds, the reversible anodic wave around +0.97 V is assigned to the (Bodipy/Bodipy+) couple. Note that this wave is less anodic with respect to phenyl or toluyl-substituted Bodipy's A [+1.11 (60) V and −1.26 (70) V versus SCE] [39]. This likely reflects the fact that the trialkoxy-gallate substituents are better electron-donating groups compared to the other derivatives. There is no indication of oxidation to the (Bodipy2+) dication within the given electrochemical window, as previously observed for pyridine linked Bodipy's [39]. The second oxidation is irreversible around +1.52 V and is likely localized on the trialkoxyphenyl subunit in keeping with literature data [40,41] and the electrochemistry of the reference compound B (+1.55 V irrev).
The single reduction is attributed to the Bodipy radical anion (Bodipy−) which was in all cases reversible and more cathodic compared to the toluyl compound A. This observation is also in keeping with the increase of electron density imported by the trialkoxyphenyl fragments.
Note that the LUMO–HOMO gap (at about 2.30 eV) remains similar along the series, as reflected by the constancy of the emission wavelength (Table 1). Changing the substitution position or increasing the number of amido fragments does not significantly change the redox potentials of the concerned fragments (Fig. 2). These results clearly reflect the combined effects of electron donating and charge delocalization and are in keeping with previous observations on related molecules [32].
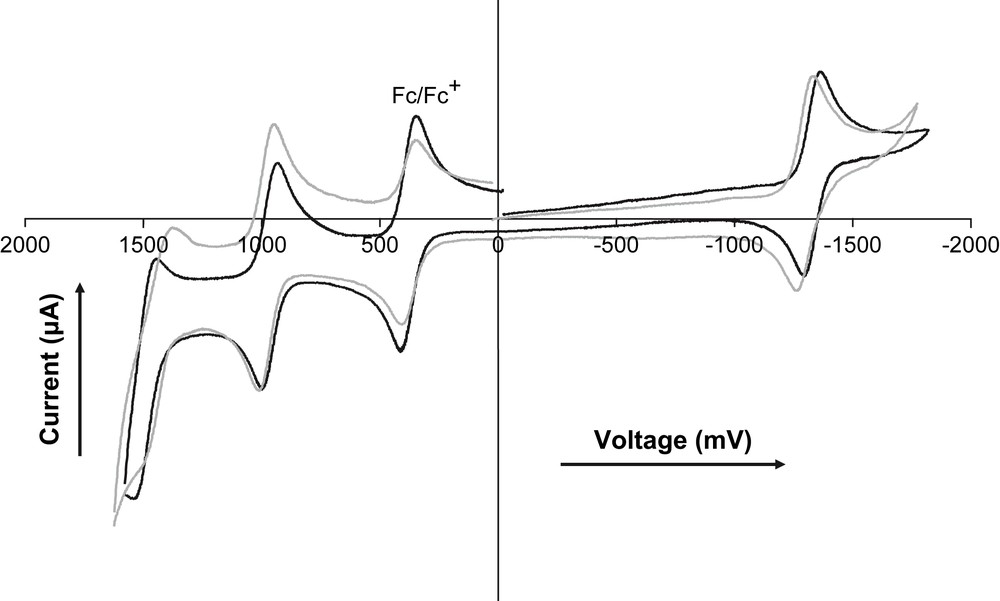
Cyclic voltammetry of compounds 5 (black) and 11 (grey) in CH2Cl2 at rt using 0.1 M nBu4PF6 as supporting electrolyte at a scan rate of 200 mV/s.
2.4 X-ray crystal structure studies
Single crystals of compounds 2a, 2b, 4a and 5 suitable for X-ray structure determinations were obtained by slow evaporation of a CH2Cl2/hexane solvent mixture. The crystal structure of compound 2b, lacking the methyl group on the dinitrophenyl fragment, has been briefly described in the literature [29], but is discussed further here to make some geometrical and structural comparisons.
2.4.1 Compound 2b
Purple parallelepiped crystals were obtained as a pure single phase crystallizing in the orthorhombic space group Pbca (a = 13.345(3) Å; b = 8.431(4) Å; c = 41.094(4) Å; V = 4624(2) Å3, Z = 8). The asymmetric unit contains one molecule, for which Fig. 3 shows the atomic numbering and labeling scheme. The molecule has effective 2-fold rotational symmetry. The Bodipy core is planar and the dinitrophenyl fragment is almost orthogonal (dihedral angle 83.6°), preventing any extended conjugation over the whole molecule. The average B–N and B–F bond lengths are 1.541(3) and 1.384(3) Å, respectively, and the N1–B–N2, F1–B–F2, N1–B–F2, N2–B–F1, N1–B–F1, F1–B–N2 angles are 107.4(2)°, 110.0(2)°, 110.2(2)°, 109.6(2)°, 110.1(2)° and 109.6(2)°, respectively. One of the two nitro groups is essentially coplanar with the phenyl ring (the dihedral angle between the mean phenyl ring and the nitro group planes is 0.91°), whereas the second is twisted by 20.86°.
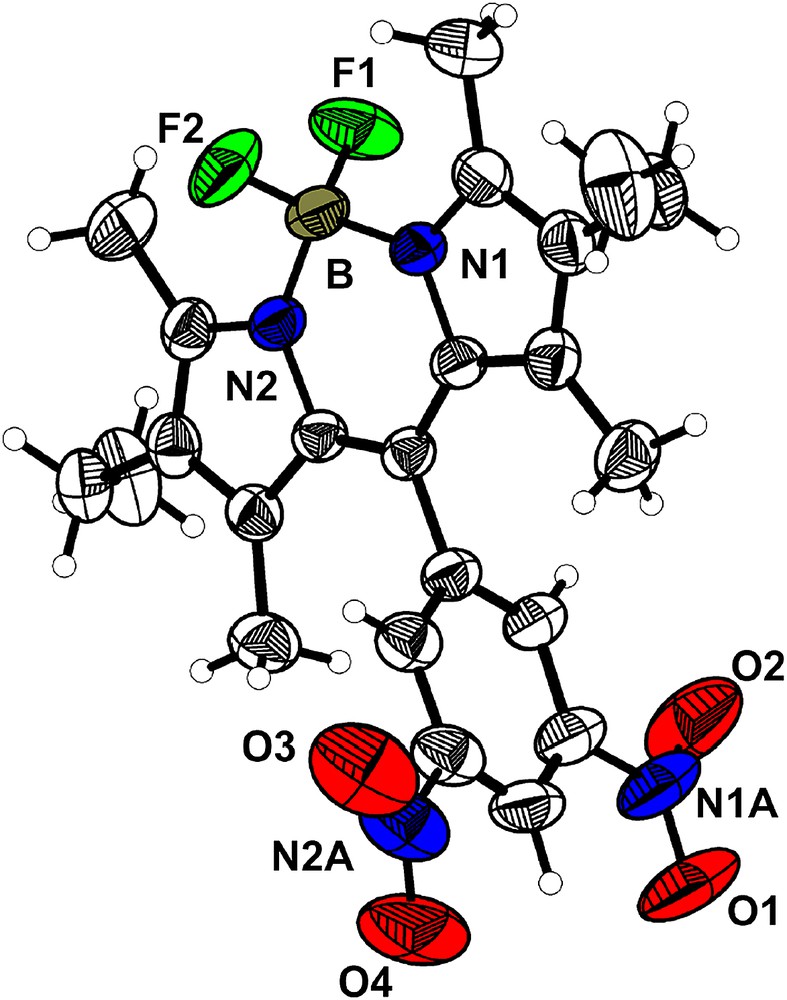
ORTEP drawing of the molecule 2b with the main numbering scheme. Thermal ellipsoids drawn at the 50% probability level.
Projection along the b axis reveals a segregation between the Bodipy cores and the dinitrophenyl moieties in the [001] direction into layers (Fig. 4). No interactions between the adjacent layers of Bodipy cores especially via the fluorine atoms (F⋯F contacts) can be observed in the c direction and only one significant type of short contacts between the nitro groups of adjacent layers along the c direction is observed (N1A–O4 = 3.029(4) Å). Thus, this structure can be described as made of isolated layers of molecules 2b in the ab plane which stack along the c direction. Inside a Bodipy layer in the ab plane, neither hydrogen bonding nor π–π stacking is observed and only short contacts between the nitro fragments are observed (O2–O3 = 2.951(4) Å).

(a) Projection of the structure of compound 2b along the b axis; (b) drawing of the Bodipy layer in the ab plane (short contacts between the nitro groups are highlighted by dashed lines). Hydrogen atoms are omitted for clarity.
2.4.2 Compound 2a
This compound crystallizes in the orthorhombic space group P212121 (a = 8.822(1) Å; b = 11.374(1) Å; c = 24.463(2) Å; V = 2454.7(4) Å3, Z = 4) with one molecule in the asymmetric unit (Fig. 5). The Bodipy fragment is planar, with the dinitrophenyl subunit almost perpendicular to it (88.9°). The geometry around the boron atom is essentially tetrahedral (B–F1, 1.381(3) Å; B–F2, 1.382(3) Å; B–N1, 1.554(3) Å; B–N2, 1.552(3) Å; N1–B–F1, 109.8(2)°; N1–B–F2, 109.9(2)°; N2–B–N1, 106.8(2)°; N2–B–F2, 109.7(2)°; F1–B–F2, 110.1(2)°; N2–B–F1, 110.5(2)°). The presence of the methyl group (C7A) in 2a induces steric hindrance and thus larger twists of the nitro fragments from the phenyl plane compared to 2b. The dihedral angles between the mean plane of the phenyl ring and the nitro groups are 29.0° for O1–N3–O2 and 67.6° for O3–N4–O4.
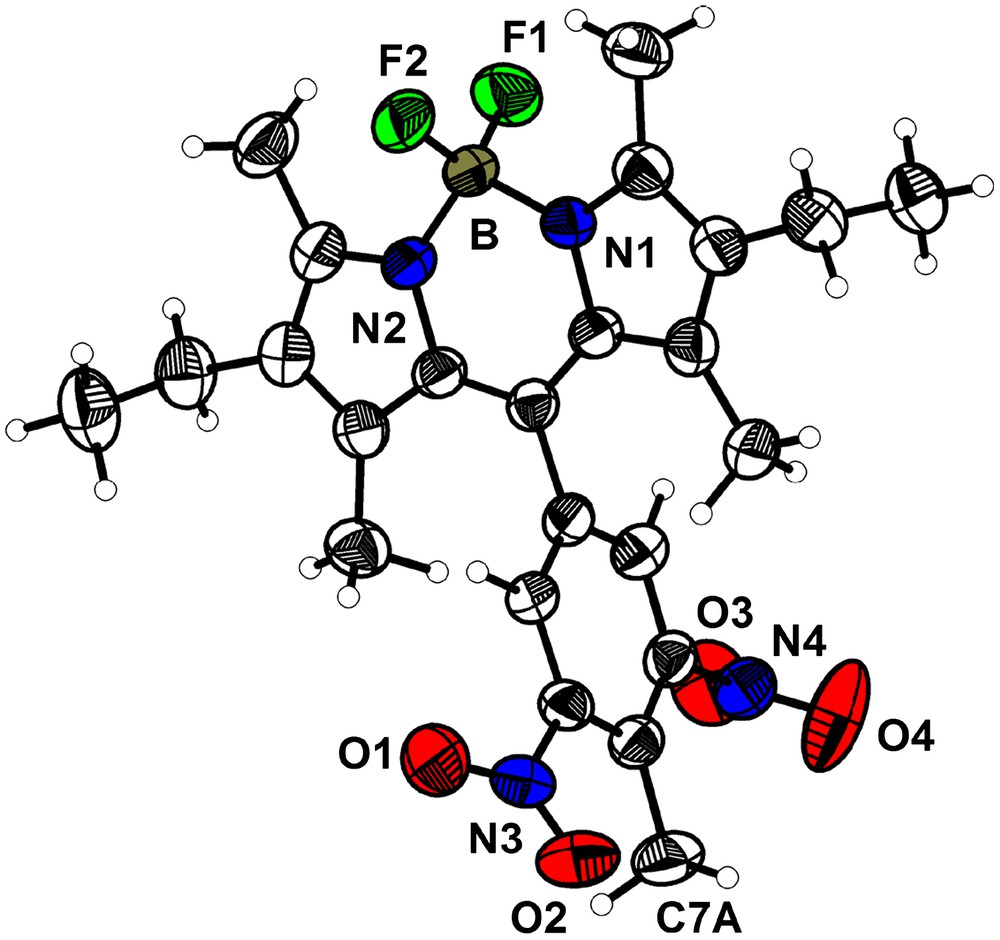
ORTEP view of the molecule 2a with the main numbering scheme (50% probability level).
Projection of the structure along the a axis shows that the lattice can be considered as made up of layers in which the Bodipy units adopt alternating orientations such that the nitro substituents lie to both sides of the layers (Fig. 6a), unlike the double layer form which results from parallel orientations of the layer constituents found in the previous structure (Fig. 4). This structure can be described as formed by single layers of Bodipy stacked in the c direction separated by the nitrophenyl groups co-facially arranged along the (011) direction. The shortest distance between the carbon of the methyl group C7A of one molecule and the centre of the nearest phenyl ring is ∼5.4 Å, but there are contacts between oxygen and nitrogen atoms of contiguous nitro groups (O3–N3 = 3.172(4); O3–O1 = 3.298(4) Å). The Bodipy layer parallel to the ab plane is depicted in Fig. 6b. No π–π stacking is apparent between the molecules inside the layers, but short contacts between fluorine atoms and nitrogen atoms are observed (2.858 Å F2–N3).
(a) Representation of the crystalline structure of compound 2a projected along the a axis; (b) view of a single layer of molecule in the ab plane (short N–F contacts are highlighted by dashed lines).
2.4.3 Compound 4a
This compound crystallizes in the monoclinic space group P21/c (a = 7.697(1) Å; b = 12.514(1) Å; c = 23.937(2) Å; β = 94.084(2)°; V = 2299.7(5) Å3, Z = 4). The molecular structure of 4a is shown in Fig. 7. The aromatic Bodipy core is planar and the pendant diaminophenyl fragment is almost perpendicular to it (dihedral angle 85.5°). The geometry around the boron atom is tetrahedral and the observed distances and angles compare well with the ones usually observed on similar compounds [42] (B1–F1, 1.380(3) Å; B1–F2, 1.393(3) Å; B1–N1, 1.530(3) Å; B1–N2, 1.535(3) Å; N1–B1–F1, 110.2(2)°; N1–B1–F2, 109.8(2)°; N2–B1–N1, 107. 9(2)°; N2–B1–F2, 109.4(2)°; F1–B1–F2, 109.3(2)°; F1–B1–N2, 110.3(2)°). Amino groups are almost coplanar with the phenyl ring (the dihedral angles being 4.6° with N1A and 10.3° with N2A).

ORTEP view of the molecule 4a with its main numbering scheme (thermal ellipsoids are at 50% probability).
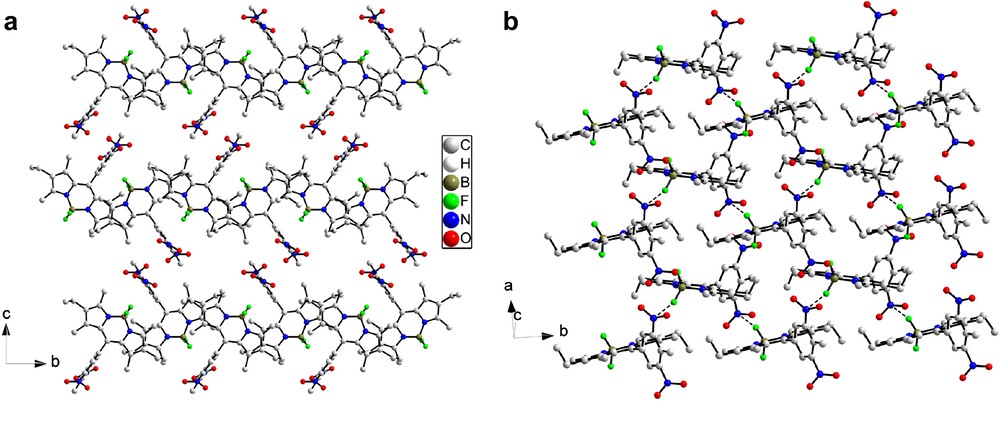
Reduction of the two nitro groups of 2a (orthorhombic, P212121) produces major changes in the crystal structure found for 4a (monoclinic, P21/c). Projected along a, the structure can be seen as a close packed arrangement of stacks containing dimers of molecules (Fig. 8). Closer examination shows that the molecules are paired head to tail with short contacts between the nitrogen atom of the amine function N1A and the fluorine atom F1 of the BF2 fragments and the N1 nitrogen atom of the dipyrromethene fragments (N1–N1A, 3.366(3) Å; F1–N1A, 3.444(3) Å). These dimers then stack in the [100] direction to form columns, but no significant interactions along the stacking direction are observed between the dimers. Short inter-stack contacts between the fluorine atoms and the methyl carbons of the phenyl fragment or of the Bodipy core (F1–C1B, 3.124(3) Å; F2–C7A, 3.253(3) Å) are also observed (Fig. 8b). Short nitrogen–nitrogen contacts (N2A–N2, 3.212(3) Å) between the amino groups and the adjacent dipyrromethene cores also occur. These additional short interactions further stabilize the edifice in the b and c directions.
(a) Stack of head to tail dimers of molecule 4a viewed along the a direction. Short contacts are highlighted as dashed lines; (b) general view of the molecular structure projected along the a direction.
2.4.4 Compound 5
This compound crystallizes in the monoclinic space group P21/c (a = 15.036(1) Å; b = 21.641(2) Å; c = 10.101(1) Å; V = 3145.8(5) Å3, Z = 4). Fig. 9 shows the atomic numbering and the labeling scheme. The geometry around the boron atom is tetrahedral with distances being B–F1, 1.382(4) Å; B–F2, 1.388(4) Å; B–N1, 1.540(4) Å; B–N2, 1.549(4) Å and angles being N1–B–F1, 110.5(3)°; N1–B–F2, 110.4(2)°; N2–B–N1, 106.8(2)°; N2–B–F2, 109.6(3)°; F1–B–F2, 109.0(3)° and N2–B–F1, 110.4(3)°. The dipyrromethene subunit (including the two pyrrole rings, the four methyl and the two methylene carbons) is planar. The central phenyl ring Ar1 is almost orthogonal to the Bodipy fragment (angle Ar1–Bodipy = 80.0°) and twisted by 39.1° from the plane of the terminal phenyl ring Ar2. The twist between the Bodipy fragment and the terminal phenyl ring Ar2 is 42.2°.
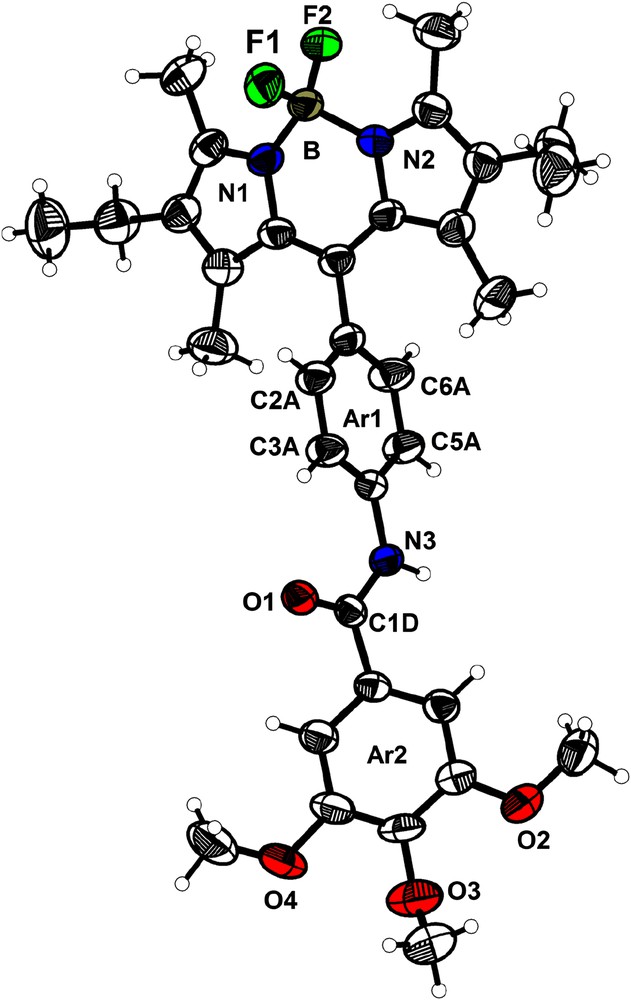
ORTEP view of the molecule 5 with principal atomic numbering (thermal ellipsoids at 50% probability level).
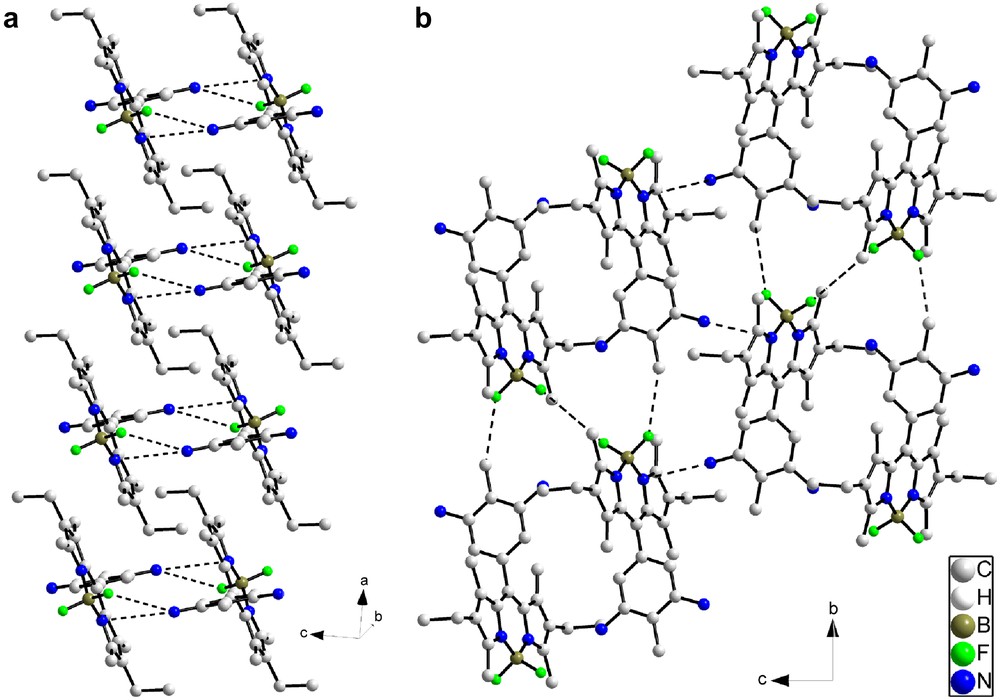
Different views of the crystal packing are presented in Figs. 10 and 11. The molecules are connected by a uniform, unidimensional hydrogen-bonded network running along the c axis (represented by dashed lines in Fig. 10). These intermolecular hydrogen bonds involve the hydrogen atom on N3 on one molecule and the oxygen atom O1 of the adjacent molecule. In this way, all the molecules are engaged in two hydrogen bonds pointing in opposite directions (N3H–O1 = 2.19 Å and N3–H–O1 angle = 155.7°) (Fig. 10a). The geometry around the boron atom is tetrahedral and the observed distances and angles compare well with the ones usually observed on similar compounds [42]. FT-IR spectroscopy is a powerful tool to probe hydrogen bonding [43,44] and, at room temperature, this compound has its amide group involved in strong hydrogen bonding, as clearly evidenced by the νNH and νCO stretching vibrations at 3308 and 1650 cm−1, respectively. Note that corresponding values for free amides usually lie at 3500–3400 cm−1 for νNH and around 1680 cm−1 for νCO [28,29]. The presence of single νNH and νCO stretching bands in IR spectra confirms a well-organized hydrogen-bonded network in which only one type of hydrogen bond is effective. The structural determination shows that this supramolecular hydrogen-bonded edifice adopts a V-shape. These hydrogen-bonded strands are further connected via the Bodipy fragments by H⋯F interactions (Fig. 10b), which results in strongly stabilized zigzag layers (Fig. 11). Closer examination of the structure reveals that the Bodipy fragments are dimerized in a centrosymmetric fashion by formation of two hydrogen bonds between one fluorine atom of the BF2 fragments (F1) on the first molecule and two hydrogen atoms on the phenyl ring Ar1 of the second molecule (C6AH⋯F1 = 2.618 Å, C6AHF1 angle = 117.3°; C5AH⋯F1 = 2.502 Å, C6AHF1 angle = 122.0°) (Fig. 10; bold dashed lines). These Bodipy dimers are further connected by weaker H⋯F interactions along the c direction (C2AH⋯F2 = 3.200 Å, C2AHF2 angle = 98.3°; C3AH⋯F2 = 3.375 Å, C3AHF2 angle = 102.2°) (thin dashed lines). The formation of Bodipy dimers orients the ethyl groups of one fluorophore in the same direction. No strong interactions are apparent between the different zigzag layers.
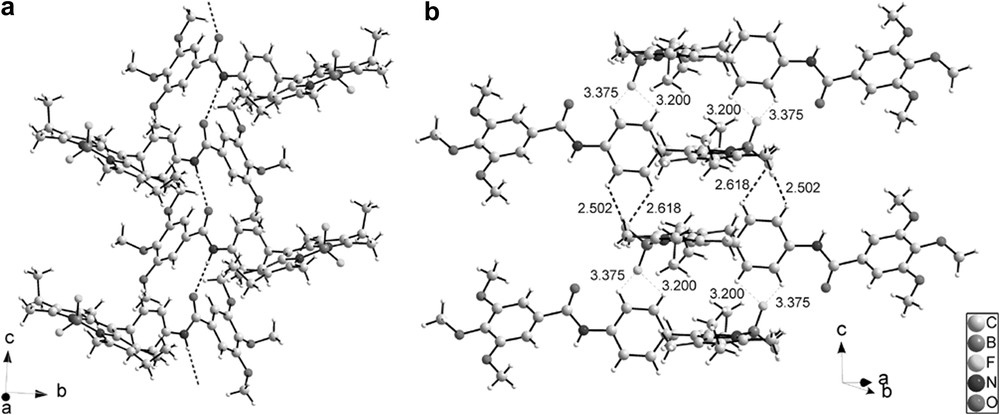
(a) View of the one-dimensional hydrogen-bonded network, represented by dotted lines, running along the c axis (N3H–O1 = 2.191(2) Å and N3–H–O1 angle = 155.63(14)°); (b) HF bonds observed in intra and inter Bodipy dimers. Strong interactions are represented as bold dotted lines and weak interactions by thin dotted lines.
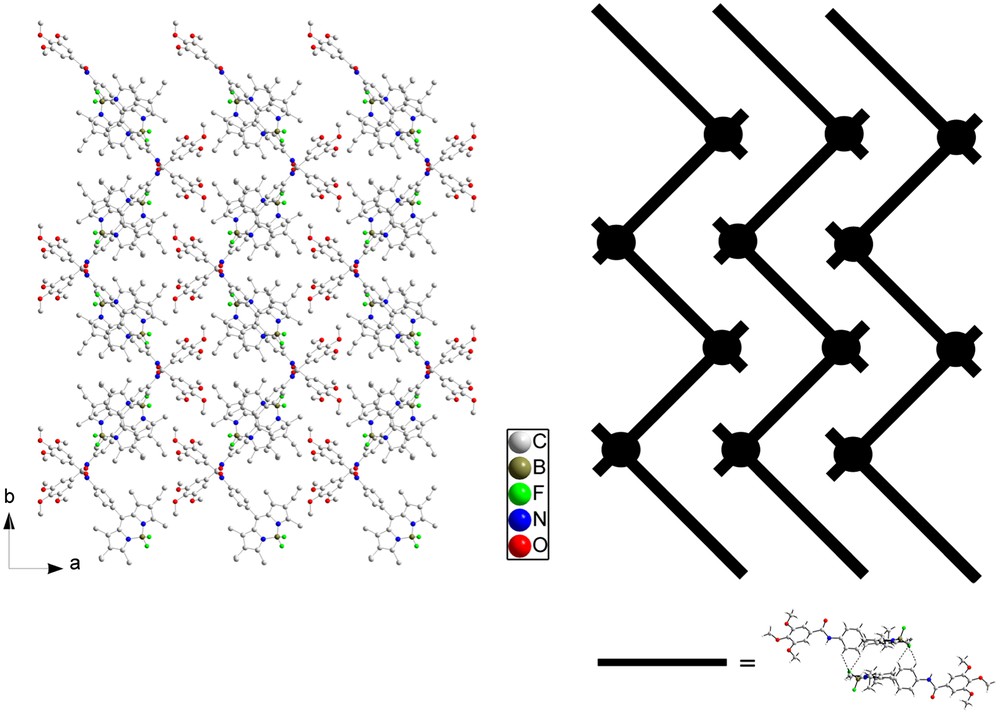
Projection of the crystalline structure of 5 along the c axis (left, hydrogen atoms omitted for clarity) and schematic representation of this structure (right). A black rod corresponds to a Bodipy dimer and a black circle to a one-dimensional hydrogen-bonded network viewed from the top.
2.5 Electroluminescence applications
These Bodipy molecules exhibit remarkable opto-electronic properties, with strong absorption in the visible region, high fluorescence quantum yields, and narrow emission bandwidths with high peak intensities. These optical properties render them attractive as organic emitters. Moreover, these neutral molecules which are thermally stable and soluble in common organic solvents can be easily deposited on a surface by vacuum evaporation or spin coating. Furthermore, the property of forming well-organized thin films on a surface may lead to improved emission efficiency in OLED applications. Additionally, these molecules are redox active, undergoing oxidation and reduction in a convenient potential range (Fig. 2), raising the prospect of their use in electroluminescence [14,45] and charge-transport devices [15]. For these reasons, compound 5, with methoxy terminal groups, has been studied in OLEDs as pure compound. The thin films were prepared by vacuum evaporation.
To evaluate the capability to transport charges and to form excitons, a simple OLED was elaborated using a pure layer of compound 5 (100 nm) sublimed onto an ITO-coated glass substrate, and using an aluminium cathode as contact electrode. The photoluminescence spectrum of the organic film presents two maxima at 545 and 633 nm upon excitation at 390 nm. The first emission peak can be safely attributed, in regard to the results obtained in solution, to the emission of monomeric and isolated Bodipy molecules. The second emission peak at 633 nm is attributed to the formation of aggregated species. In fact, it has been recently demonstrated that Bodipy molecules can aggregate to form dimer or larger aggregates emitting at lower energies [28,29]. The excitation spectra for the two emissions are identical and match the absorption spectra over the entire spectral range.
Electroluminescence spectra were collected for various applied voltages on this simple ITO/Bodipy(5) (100 nm)/Al OLED configuration. Interestingly, the electroluminescence spectra also displayed two intense emission peaks at 550 nm (yellow) and 635 nm (red) (Fig. 12), very similar to the photoluminescence spectra measured on the same device. Upon increasing the applied voltage, the electroluminescence intensity increases, but the relative intensity of the two emission peaks remains constant. This dual emission in the yellow and in the red part of the visible spectrum explains the orange colour detected with the spectroradiometer. Clearly, this compound can emit when simply sandwiched between ITO-coated glass and aluminium electrodes. Compounds with long carbon chains have also been tested but, when a potential is applied, the compounds start to melt and the formation of collapsed droplets can be observed by optical microscopy, meaning the OLED is unstable.
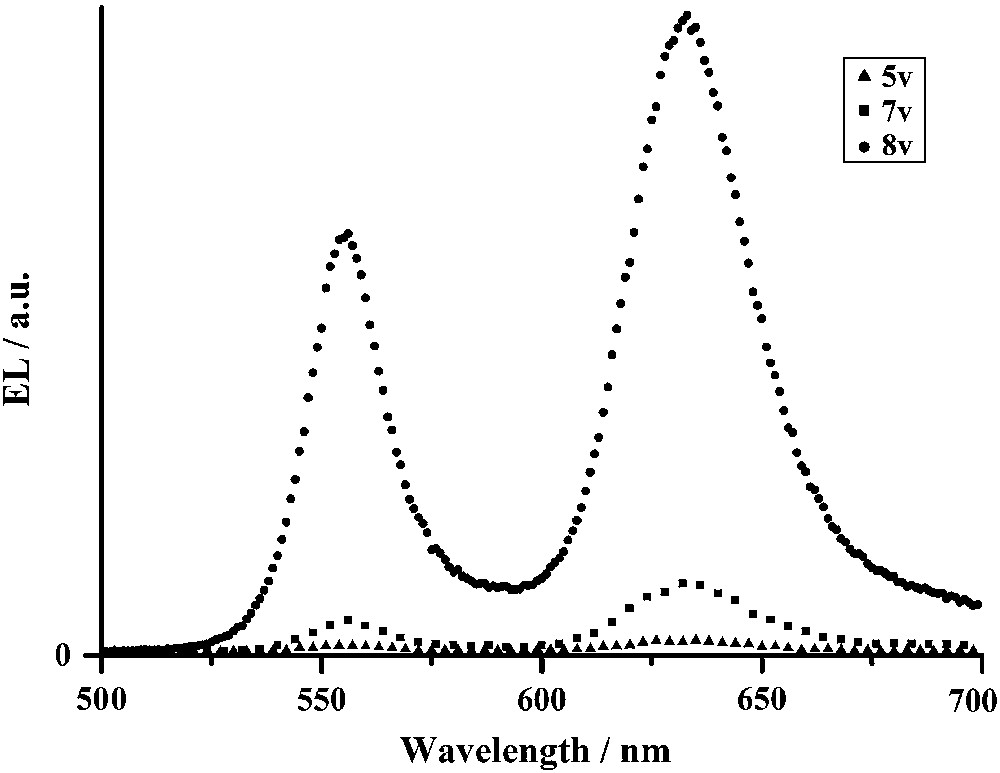
Electroluminescence spectra of an OLED structure ITO/Bodipy(5) (100 nm)/Al for various applied voltages.
Interestingly, by dispersing compound 5 in a PVK polymer matrix by spin coating, solely yellow emission has also been observed with the structure ITO/PEDOT:PSS (60 nm)/PVK:Bodipy (10 wt%, 100 nm)/Al (Fig. 13). This result obtained with a dopant concentration of ∼3 mol% is in keeping with the absence of Bodipy aggregation at that concentration level. Here it is assumed in light of the spectral overlap between the Bodipy absorption (Fig. 1) and the PVK emission at 410 nm [46], that complete energy transfer from the PVK to the second S0 → S2 transition of the Bodipy is occurring. Previous studies in solution have proven that this process is as efficient as an overlapping with the S0 → S1 transition of the Bodipy [47].
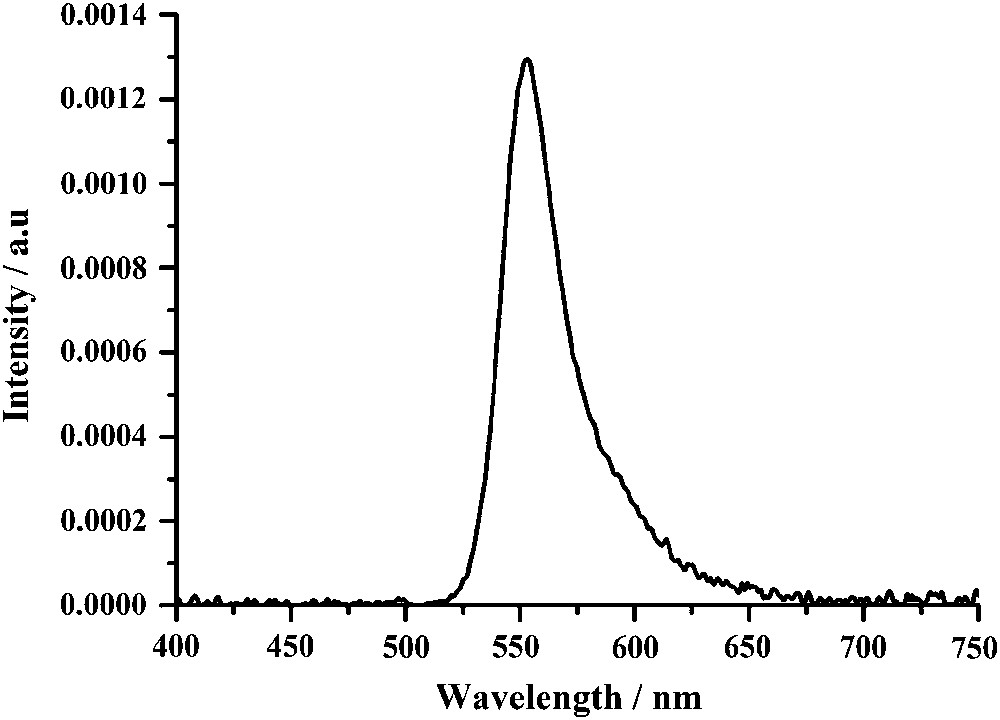
Electroluminescence spectrum of the OLED structure ITO/PEDOT:PSS (60 nm)/PVK:Bodipy(5) (10 wt%, 100 nm)/Al.
3 Conclusion
Due to the propensity of functionalized Bodipy dyes to form aggregates in the solid state as demonstrated in the crystal packing of several of these new dyes, thin films incorporating these materials show emissions in the yellow and red due to monomer and aggregates, respectively. It is foreseen that a fine control of the doping rate will allow a fine tuning of the emitted light from yellow to red. This unusual tuning is only observed in the solid state. By dispersing the Bodipy dye into a PVK polymer by spin coating, yellow light is solely observed in the OLED. Complete energy transfer from the PVK to the Bodipy is occurring, involving in this case the S0 → S2 transition of the Bodipy. Work in progress concerns the addition of polymerisable groups at the end of the long paraffin chains in order to better control the stacking of the molecules and to avoid the demixing problems found with the long carbon chains.
4 Experimental section
NMR spectra (300.1 MHz (1H) and 75.5 MHz (13C)) were recorded at room temperature using perdeuterated solvents as internal standards: ‰ (H) in parts per million relative to residual protiated solvent; ‰ (C) in parts per million relative to the solvent. A ZAB–HF–VB-analytical apparatus in FAB+ mode was used with m-nitrobenzyl alcohol (m-NBA) as matrix. Chromatographic purification was conducted using 40–63 μm silica gel. Thin layer chromatography (TLC) was performed on silica gel plates coated with fluorescent indicator. FT-IR spectra were recorded using a PerkinElmer “spectrum one” spectrometer as thin films deposited onto dry KBr pellets. UV–vis spectra were recorded using a UVIKON 940/941 dual-beam grating spectrophotometer (Kontron Instruments) with a 1-cm quartz cell. Fluorescence spectra were recorded on a PerkinElmer LS50B spectrofluorimeter. All fluorescence spectra were corrected. The fluorescence quantum yield (Φexp) was calculated from Eq. (1). Here, F denotes the integral of the corrected fluorescence spectrum, A is the absorbance at the excitation wavelength, and n is the refractive index of the medium. The reference system used was rhodamine 6G (Φref = 0.78, λexc = 488 nm) [25] in air-equilibrated water.
| (1) |
Luminescence lifetimes were measured on a PTI QuantaMaster spectrofluorimeter, using TimeMaster software with Time-Correlated Single Photon Mode coupled to a stroboscopic system. The excitation source was a thyratron-gated flash lamp filled with nitrogen gas. No filter was used for the excitation. An interference filter centred at 550 nm selected the emission wavelengths. The instrument response function was determined by using a light-scattering solution (LUDOX). Crystal data, intensity measurements, and structure refinement for all crystallized compounds are given in Table 2.
Summary of crystal data, intensity measurements and structure refinement for all crystallized compounds
| Compound 2a | Compound 2b | Compound 4 | Compound 5 | |
| Formula | C24 H27 B F2 N4 O4 | C23 H25 B F2 N4 O4 | C24 H31 B F2 N4 | C33 H38 B F2 N3 O4 |
| Mr | 484.31 | 470.28 | 424.34 | 589.47 |
| Crystal system | Orthorhombic | Orthorhombic | Monoclinic | Monoclinic |
| Space group | P212121 | Pbca | P21/c | P21/c |
| a [Å] | 8.822(1) | 13.345(3) | 7.697(1) | 15.036(1) |
| b [Å] | 11.374(1) | 8.431(4) | 12.514 (1) | 21.640(2) |
| c [Å] | 24.463(2) | 41.094(4) | 23.937(2) | 10.101(1) |
| α [°] | 90.00 | 90.00 | 90.00 | 90.00 |
| β [°] | 90.00 | 90.00 | 94.084(2) | 106.837(2) |
| γ [°] | 90.00 | 90.00 | 90.00 | 90.00 |
| V [Å3] | 2454.7(4) | 4624(2) | 2299.7(5) | 3145.8(5) |
| Z, Z′ | 4, 1 | 8, 1 | 4, 1 | 4, 1 |
| F(000) | 1016 | 1968 | 904 | 1248 |
| ρcalcd [Mg/m3] | 1.311 | 1.351 | 1.226 | 1.245 |
| μ [mm−1] | 0.100 | 0.087 | 0.084 | 0.089 |
| Crystallization method | Slow evaporation in a CH2Cl2/hexane mixture | |||
| Crystal description | Red prism | Purple thin plate | Red prism | Red thin plate |
| Crystal size [mm] | 0.40 × 0.30 × 0.20 | 0.55 × 0.30 × 0.15 | 0.50 × 0.20 × 0.20 | 0.35 × 0.35 × 0.05 |
| Diffractometer | Nonius kappa CCD | Enraf–Nonius CAD4 | Nonius kappa CCD | Nonius kappa CCD |
| λ [Å] | 0.71073 | 1.54180 | 0.71073 | 0.71073 |
| Data collection strategy | ϕ and ω scans | non-profiled ω/2θ scans | ϕ and ω scans | ϕ and ω scans |
| T [K] | 293(2) | 293(2) | 293(2) | 293(2) |
| Reflections collected/unique | 16,624/2506 | 7816/4109 | 109,76/3517 | 42,252/3609 |
| θ range [°] (completeness) | 2.45 < θ < 5.23 (98.2%) | 2.15 < θ < 67.15 (99.6%) | 1.84 < θ < 23.82 (99.2%) | 1.88 < θ < 21.76 (96.3%) |
| hkl range | −10 ≤ h ≤ 10 | −15 ≤ h ≤ 3 | −8 ≤ h ≤ 8 | −15 ≤ h ≤ 15 |
| −13 ≤ k ≤ 13 | −3 ≤ k ≤ 10 | −14 ≤ k ≤ 13 | −21 ≤ k ≤ 22 | |
| −29 ≤ l ≤ 29 | −2 ≤ l ≤ 49 | −27 ≤ l ≤ 27 | −10 ≤ l ≤ 10 | |
| Absorption correction | Multi-scan | Multi-scan | Multi-scan | Multi-scan |
| Tmax/Tmin | 0.980/0.890 | 0.878/0.772 | 0.985/0.898 | 0.991/0.974 |
| Refinement method | Least squares on F2 | |||
| H atom treatment | H-atom located from Fourier syntheses but parameters constrained with a riding model | |||
| Refined data/I > 2σ(I) | 2506/2212 | 4109/2958 | 3517/2431 | 3609/2815 |
| Restraints/parameters | 0/323 | 0/312 | 0/287 | 0/397 |
| Goodness-of-fit on F2 | 1.044 | 1.030 | 1.031 | 1.062 |
| Final R indices [I > 2σ(I)] | R1 = 0.0382, wR2 = 0.0988 | R1 = 0.0532, wR2 = 0.1415 | R1 = 0.0502, wR2 = 0.1267 | R1 = 0.0524, wR2 = 0.1302 |
| R indices (all data) | R1 = 0.0453, wR2 = 0.1089 | R1 = 0.0779, wR2 = 0.1588 | R1 = 0.0808, wR2 = 0.1448 | R1 = 0.0732, wR2 = 0.1426 |
| Δρmax, Δρmin [e Å−3] | 0.136, −0.169 | 0.220, −0.229 | 0.176, −0.204 | 0.343, −0.247 |
| CCDC number | 639642 | 289577 | 639643 | 639641 |
Syntheses of compounds 1, 3, 2b and 4b have already been reported in the literature [32].
4.1 Synthesis of compounds 2a and 4a
4.1.1 Compound 2a
In a flame-dried Schlenk flask, under argon, oxalyl chloride (1.5 equiv, 3.32 mmol) and a catalytic amount of distilled pyridine were added to a stirred solution of 4-methyl-3,5-dinitro-benzoic acid (0.5 g, 2.21 mmol) in 30 mL of distilled CH2Cl2. The mixture was stirred for 20 h at room temperature. The solvent was evaporated, as well as the excess oxalyl chloride. 30 mL of freshly distilled CH2Cl2 were then added, and the resulting solution was transferred via cannula into a degassed solution of kryptopyrrole (2 equiv, 4.42 mmol, 0.6 mL) in 20 mL of distilled CH2Cl2. The reaction mixture was stirred at room temperature for 72 h, and slowly turned from pale brown to deep purple. Triethylamine (6 equiv, 13.3 mmol, 1.9 mL) was then added and the reaction mixture turned brown–orange. After 10 min of stirring, boron trifluoride etherate was added (8 equiv, 17.7 mmol, 2.2 mL) and the reaction mixture turned purple. The solution was stirred 48 h at room temperature. It was then washed with three portions of saturated aqueous NaHCO3 (3 × 50 mL). The organic layer was dried over MgSO4, filtered, and the solvent evaporated. The crude product was purified by chromatography on a column packed with flash silica gel, using CH2Cl2/petroleum ether (50:50 to 100:0) as eluent. Recrystallization in CH2Cl2/hexane afforded the desired compound as purple crystals (0.465 g, 43%). 1H NMR (CDCl3, 300 MHz): δ (ppm) = 7.97 (s, 2H), 2.68 (s, 3H), 2.54 (s, 6H), 2.32 (q, 4H, 3J = 7.7 Hz), 1.36 (s, 6H), 1.00 (t, 6H, 3J = 7.5 Hz); 13C NMR (CDCl3, 75 MHz): δ (ppm) = 156.2, 152.2, 137.2, 136.3, 134.3, 133.1, 130.3, 128.0, 127.5, 17.2, 15.0, 14.7, 12.8; UV–vis (CH2Cl2) λ (nm) (ɛ, M−1 cm−1) = 535 (90,000), 498 (sh, 32,000), 382 (17,000); IR (KBr, cm−1): ν = 2961, 2925, 2868, 1540, 1470, 1411, 1347, 1314, 1271, 1253, 1180, 1157, 1114, 1101, 1061, 1036, 972; FAB+-MS m/z (nature of peak, relative intensity): 485.2 ([M + H]+, 100), 465.2 ([M − F]+, <10); Anal. Calcd for C24H27BF2N4O4: C, 59.52; H, 5.62; N, 11.57. Found: C, 59.26; H, 5.43; N, 11.31.
4.1.2 Compound 4a
Under argon, 5% Pd/C (0.210 g) was added to a stirred, degassed CH2Cl2/EtOH (20 mL/20 mL) solution of 2 (0.415 g, 0.87 mmol). The reaction mixture was stirred under H2 for 24 h, when residual 2 could not be detected by TLC (SiO2; CH2Cl2). The solution was then filtered through Celite and the solvents evaporated. The crude product was purified by chromatography on a column packed with flash silica gel, using CH2Cl2/petroleum ether (80:20–100:0). Recrystallization in CH2Cl2/hexane afforded the desired compound as an orange powder (0.228 g, 64%). 1H NMR (CDCl3, 300 MHz): δ (ppm) = 6.10 (s, 2H), 3.63 (br s, 4H), 2.51 (s, 6H), 2.31 (q, 4H, 3J = 7.5 Hz), 2.05 (s, 3H), 1.54 (s, 6H), 0.99 (t, 6H, 3J = 7.5 Hz); 13C NMR (CDCl3, 75 MHz): δ (ppm) = 153.2, 146.0, 141.2, 138.8, 134.2, 132.5, 130.8, 107.2, 106.4, 17.2, 14.8, 12.6, 11.8, 10.5; UV–vis (CH2Cl2) λ (nm) (ɛ, M−1 cm−1) = 523 (90,000), 492 (sh, 30,000), 372 (15,000); IR (KBr, cm−1): ν = 2973, 2962, 1627, 1576, 1540, 1476, 1320, 1263, 1192, 1161, 1115, 1061, 978; FAB+-MS m/z (nature of peak, relative intensity): 425.2 ([M + H]+, 100), 405.2 ([M − F]+, 20); Anal. Calcd for C24H31BF2N4: C, 67.93; H, 7.36; N, 13.20. Found: C, 67.67; H, 7.55; N, 13.55.
4.2 General procedure for the synthesis of compounds 5–10
A stirred solution of 3,4,5-tris-alkyloxy-benzoic acid in freshly distilled thionyl chloride was refluxed for 3 h, the excess of SOCl2 was then evaporated off and the resulting solid dried under vacuum for 3 h to obtain the benzoic acid chloride, which was used without any further purification.
Compound 3 and triethylamine (2 equiv) were added to a stirred solution of the benzoic acid chloride (1 equiv) in distilled CH2Cl2. The resulting mixture was stirred at room temperature for 12 h, when total consumption of the starting material was observed. The reaction mixture was then washed with water, the organic layer dried over anhydrous MgSO4, and the solvent evaporated. Purification was performed using a chromatography column packed with silica gel, using CH2Cl2/petroleum ether and CH2Cl2/MeOH as eluents and then recrystallized from CH2Cl2/CH3CN, CH2Cl2/MeOH or CH2Cl2/hexane.
4.2.1 Compound 5
The compound was prepared from 0.072 g of 3,4,5-tris-methoxy-benzoic acid (0.34 mmol), 3 mL of SOCl2, 0.0134 g of 3 (0.34 mmol), 0.09 mL of distilled triethylamine (0.68 mmol) and 20 mL of distilled CH2Cl2 to give 0.181 g of 7 after crystallization from CH2Cl2/hexane (90%). 1H NMR (CDCl3, 300 MHz): δ (ppm) = 7.90 (s, 1H), 7.79 (dd, 2H, 3J = 8.5 Hz, 4J = 1.7 Hz), 7.29 (dd, 2H, 3J = 8.7 Hz, 4J = 1.9 Hz), 7.11 (s, 2H), 3.94 (s, 6H), 3.92 (s, 3H), 2.53 (s, 6H), 2.30 (q, 4H, 3J = 7.5 Hz), 1.36 (s, 6H), 0.98 (t, 6H, 3J = 7.5 Hz); 13C NMR (CDCl3, 75 MHz): δ (ppm) = 165.7, 153.6, 141.6, 139.7, 138.7, 138.4, 133.0, 131.9, 131.1, 130.3, 129.3, 120.5, 104.7, 61.1, 56.6, 17.2, 14.8, 12.7, 12.1; 11B NMR (CDCl3, 128 MHz): δ (ppm) = 3.87 (t, 1J = 32 Hz); UV–vis (CH2Cl2) λ (nm) (ɛ, M−1 cm−1) = 525 (60,000), 492 (sh, 19,000), 367 (9400), 276 (24,000); IR (KBr, cm−1): ν = 2960, 2932, 2870, 2839, 1675, 1648, 1583, 1539, 1497, 1474, 1413, 1399, 1373, 1363, 1334, 1318, 1295, 1273, 1235, 1184, 1124, 1072, 1045, 1007, 977; FAB+-MS m/z (nature of peak, relative intensity): 590.2 ([M + H]+, 100), 570.2 ([M − F]+, 15); Anal. Calcd for C33H38BF2N3O4: C, 67.24; H, 6.50; N, 7.13. Found: C, 67.01; H, 6.27; N, 6.86.
4.2.2 Compound 6
The compound was prepared from 0.115 g of 3,4,5-tris-octyloxy-benzoic acid (0.23 mmol), 3 mL of SOCl2, 0.090 g of 3 (0.23 mmol), 0.06 mL of distilled triethylamine (0.46 mmol) and 20 mL of distilled CH2Cl2 to give 0.122 g of 6 after precipitation from CH2Cl2/CH3CN (61%). 1H NMR (CDCl3, 400 MHz): δ (ppm) = 7.84 (s, 1H), 7.78 (dd, 2H, 3J = 8.3 Hz, 4J = 1.6 Hz), 7.28 (dd, 2H, 3J = 8.7 Hz, 4J = 2.1 Hz), 7.08 (s, 2H), 4.07–4.02 (m, 6H, OCH2), 2.53 (s, 6H), 2.31 (q, 4H, 3J = 7.5 Hz), 1.90–1.72 (m, 6H, CH2), 1.53–1.44 (m, 6H, CH2), 1.41–1.20 (m, 30H, CH3 + CH2), 0.99 (t, 6H, 3J = 7.4 Hz), 0.92–0.85 (m, 9H, CH3); 13C NMR (CDCl3, 75 MHz): δ (ppm) = 165.9, 153.9, 153.5, 142.0, 139.8, 138.8, 138.5, 133.0, 131.8, 131.1, 129.8, 129.3, 120.4, 106.1, 73.7, 69.7, 32.04, 32.0, 31.0, 30.5, 29.7, 29.53, 29.51, 29.4, 26.2, 22.84, 22.81, 17.2, 14.7, 14.2, 12.6, 12.1; 11B NMR (CDCl3, 128 MHz): δ (ppm) = 3.87 (t, 1J = 32 Hz); UV–vis (CH2Cl2) λ (nm) (ɛ, M−1 cm−1) = 525 (73,000), 493 (sh, 24,000), 367 (12,000), 278 (32,000); IR (KBr, cm−1): ν = 2971, 2926, 2856, 1649, 1582, 1547, 1512, 1495, 1398, 1337, 1264, 1238, 1114, 1084, 983; FAB+-MS m/z (nature of peak, relative intensity): 884.1 ([M + H]+, 100), 864.2 ([M − F]+, 20); Anal. Calcd for C54H80BF2N3O4: C, 73.37; H, 9.12; N, 4.75. Found: C, 73.17; H, 8.75; N, 4.42.
4.2.3 Compound 7
The compound was prepared from 0.128 g of 3,4,5-tris-dodecyloxy-benzoic acid (0.19 mmol), 3 mL of SOCl2, 0.075 g of 3 (0.19 mmol), 0.05 mL of distilled triethylamine (0.38 mmol) and 20 mL of distilled CH2Cl2 to give 0.109 g of 7 after precipitation from CH2Cl2/CH3CN (55%). 1H NMR (CDCl3, 300 MHz): δ (ppm) = 7.82–7.76 (m, 3H), 7.28 (d, 2H, 3J = 8.7 Hz), 7.08 (s, 2H), 4.1–4.0 (m, 6H, OCH2), 2.53 (s, 6H), 2.31 (q, 4H, 3J = 7.5 Hz), 1.88–1.72 (m, 6H, CH2), 1.51–1.27 (m, 60H, CH2 + CH3), 0.98 (t, 6H, 3J = 7.5 Hz), 0.88 (m, 9H, CH3); 13C NMR (CDCl3, 75 MHz): δ (ppm) = 165.8, 157.9, 153.4, 141.9, 136.2, 132.9, 131.1, 129.3, 120.4, 113.7, 107.8, 106.0, 31.9, 29.74, 29.71, 29.6, 29.41, 29.37, 26.1, 22.7, 17.1, 14.8, 14.6, 14.1, 12.5, 11.9; 11B NMR (CDCl3, 128 MHz): δ (ppm) = 3.86 (t, 1J = 32 Hz); UV–vis (CH2Cl2) λ (nm) (ɛ, M−1 cm−1) = 525 (72,000), 492 (sh, 24,000), 370 (13,000), 278 (32,000); IR (KBr, cm−1): ν = 2918, 2849, 1644, 1579, 1541, 1497, 1470, 1390, 1335, 1315, 1263, 1236, 1189, 1114, 1075, 976; FAB+-MS m/z (nature of peak, relative intensity): 1052.1 ([M + H]+, 100), 1032.1 ([M − F]+, 10); Anal. Calcd for C66H104BF2N3O4: C, 75.33; H, 9.96; N, 3.99. Found: C, 75.18; H, 9.64; N, 3.64.
4.2.4 Compound 8
The compound was prepared from 0.139 g of 3,4,5-tris-hexadecyloxy-benzoic acid (0.16 mmol), 3 mL of SOCl2, 0.065 g of 3 (0.16 mmol), 0.05 mL of distilled triethylamine (0.32 mmol) and 20 mL of distilled CH2Cl2 to give 0.135 g of 8 after precipitation from CH2Cl2/CH3CN (89%). 1H NMR (CDCl3, 300 MHz): δ (ppm) = 7.84–7.76 (m, 3H), 7.28 (d, 2H, 3J = 8.5 Hz), 7.08 (s, 2H), 4.1–4.0 (m, 6H, OCH2), 2.53 (s, 6H), 2.31 (q, 4H, 3J = 7.5 Hz), 1.88–1.71 (m, 6H, CH2), 1.49–1.26 (m, 84H, CH2 + CH3), 0.99 (t, 6H, 3J = 7.5 Hz), 0.88 (m, 9H, CH3); 13C NMR (CDCl3, 75 MHz): δ (ppm) = 165.9, 153.9, 153.5, 145.2, 143.2, 142.0, 139.7, 138.8, 138.4, 132.9, 131.8, 131.1, 129.8, 129.3, 120.3, 106.0, 73.7, 69.7, 32.1, 30.5, 29.9, 29.8, 29.7, 29.6, 29.5, 26.2, 22.8, 17.2, 14.8, 14.2, 12.7, 12.1; 11B NMR (CDCl3, 128 MHz): δ (ppm) = 3.87 (t, 1J = 32 Hz); UV–vis (CH2Cl2) λ (nm) (ɛ, M−1 cm−1) = 525 (77,000), 493 (sh, 30,000), 362 (20,000), 276 (40,000); IR (KBr, cm−1): ν = 3285, 2951, 2916, 2849, 1673, 1648, 1584, 1540, 1524, 1497, 1467, 1423, 1403, 1383, 1337, 1313, 1276, 1264, 1179, 1158, 1115, 1082, 1057, 969; FAB+-MS m/z (nature of peak, relative intensity): 1220.2 ([M + H]+, 100), 1200.1 ([M−F]+, 25); Anal. Calcd for C78H128BF2N3O4: C, 76.75; H, 10.57; N, 3.44. Found: C, 76.39; H, 10.27; N, 3.21.
4.2.5 Compound 9
This compound was prepared from 0.146 g of 3,4,5-tris-(3,7,11,15-tetramethyl-hexadecyloxy)-benzoic acid (0.14 mmol), 3 mL of SOCl2, 0.057 g of 3 (0.14 mmol), 0.04 mL of distilled triethylamine (0.28 mmol) and 20 mL of distilled CH2Cl2 to give 0.102 g of 9 after precipitation from CH2Cl2/CH3CN (51%). 1H NMR (CDCl3, 300 MHz): δ (ppm) = 7.84 (br s, 1H), 7.78 (d, 2H, 3J = 8.7 Hz), 7.28 (d, 2H, 3J = 8.4 Hz), 7.1 (s, 2H), 4.12–4.03 (m, 6H, OCH2), 2.53 (s, 6H), 2.31 (q, 4H, 3J = 7.5 Hz), 1.93–0.83 (m, 129H); 13C NMR (CDCl3, 75 MHz): δ (ppm) = 165.9, 153.9, 153.5, 142.0, 139.8, 138.8, 138.4, 133.0, 131.8, 131.1, 129.8, 129.3, 120.4, 106.0, 72.0, 68.1, 39.5, 37.7, 37.63, 37.59, 37.5, 37.4, 36.6, 36.5, 32.96, 30.07, 29.9, 28.1, 24.9, 24.6, 22.9, 22.8, 19.9, 19.83, 19.78, 19.7, 17.2, 14.7, 12.6, 12.1; UV–vis (CH2Cl2) λ (nm) (ɛ, M−1 cm−1) = 525 (95,000), 492 (sh, 35,000), 363 (23,000), 276 (50,000); IR (KBr, cm−1): ν = 2954, 2925, 2868, 1651, 1582, 1543, 1498, 1476, 1426, 1399, 1374, 1337, 1318, 1272, 1236, 1196, 1113, 1080, 1050, 982; FAB+-MS m/z (nature of peak, relative intensity): 1388.2 ([M + H]+, 100), 1368.2 ([M−F]+, 50); Anal. Calcd for C90H152BF2N3O4: C, 77.82; H, 11.03; N, 3.03. Found: C, 77.44; H, 10.64; N, 2.67.
4.2.6 Compound 10
The compound was prepared from 0.099 g of 3,4,5-tris-(3,7-dimethyl-octyloxy)-benzoic acid (0.17 mmol), 3 mL of SOCl2, 0.067 g of 3 (0.17 mmol), 0.05 mL of distilled triethylamine (0.34 mmol) and 20 mL of distilled CH2Cl2 to give 0.106 g of 10 after precipitation from CH2Cl2/CH3CN (63%). 1H NMR (CDCl3, 300 MHz): δ (ppm) = 7.85 (br s, 1H), 7.78 (d, 2H, 3J = 8.5 Hz), 7.28 (d, 2H, 3J = 8.5 Hz), 7.09 (s, 2H), 4.15–4.0 (m, 6H, OCH2), 2.53 (s, 6H), 2.31 (q, 4H, 3J = 7.5 Hz), 2.0–1.47 (m, 12H, CH + CH2), 1.36–1.15 (m, 24H, CH2 + CH3), 1.02–0.86 (m, 33H, CH3); 13C NMR (CDCl3, 75 MHz): δ (ppm) = 165.9, 153.9, 153.5, 142.0, 139.8, 138.8, 138.4, 133.0, 131.8, 131.1, 129.8, 129.3, 120.4, 106.0, 72.0, 68.0, 39.5, 39.4, 37.7, 37.5, 36.5, 30.0, 29.8, 28.1, 24.9, 22.85, 22.76, 22.7, 19.7, 17.2, 14.7, 14.2, 12.6, 12.1; UV–vis (CH2Cl2) λ (nm) (ɛ, M−1 cm−1) = 525 (100,000), 490 (sh, 33,000), 364 (20,000), 278 (45,000); IR (KBr, cm−1): ν = 3288, 2990, 2922, 3695, 1650, 1579, 1539, 1519, 1493, 1473, 1462, 1423, 1392, 1364, 1335, 1315, 1274, 1261, 1232, 1194, 1109, 1075, 1044, 980; FAB+-MS m/z (nature of peak, relative intensity): 968.0 ([M + H]+, 100), 948.0 ([M − F]+, 15); Anal. Calcd for C60H92BF2N3O4: C, 74.43; H, 9.58; N, 4.34. Found: C, 74.20; H, 9.35; N, 4.53.
4.3 General procedure for the synthesis of compounds 11–13
In a round-bottomed flask, under argon, to a stirred solution of 4 in distilled CH2Cl2, were sequentially added 3,4,5-tris-alkyloxy-benzoic acid (2 equiv), 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (4 equiv) and finally 2,2-dimethylaminopyridine (4 equiv). The mixture was stirred and heated to reflux until TLC indicated no more evolution of the reaction. The reaction mixture was washed with water and extracted with CH2Cl2. The organic layer was dried over MgSO4, filtered and the solvent evaporated. The crude product was purified by chromatography on a column packed with silica gel, using CH2Cl2/petroleum ether or CH2Cl2/AcOEt as eluent and then recrystallized from CH2Cl2/CH3CN.
4.3.1 Compound 11
The compound was prepared from 0.080 g of 3,4,5-tris-octyloxy-benzoic acid (0.16 mmol), 0.030 g of 4 (0.072 mmol), 0.055 g of 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (0.29 mmol), 0.035 g of 2,2-dimethylaminopyridine (0.29 mmol) and 20 mL of distilled CH2Cl2 to give 0.065 g of 11, recovered as an orange powder after precipitation (65%). 1H NMR (CDCl3, 300 MHz): δ (ppm) = 7.68 (s, 2H), 7.51 (s, 2H), 7.09 (s, 4H), 4.06–4.00 (m, 12H), 2.52 (s, 6H), 2.33–2.26 (q + s, 4 + 3H, 3J = 7.5 Hz), 1.87–1.70 (m, 12H), 1.53–1.25 (m, 66H), 0.99 (t, 6H, 3J = 7.5 Hz), 0.91–0.85 (m, 18H); 13C NMR (CDCl3, 75 MHz): δ (ppm) = 165.9, 154.0, 153.5, 142.1, 138.6, 137.4, 134.4, 132.9, 130.9, 129.1, 126.5, 122.7, 106.2, 73.8, 69.7, 32.04, 31.96, 30.5, 29.8, 29.7, 29.52, 29.49, 29.4, 26.2, 22.84, 22.81, 17.2, 14.8, 14.2, 13.3, 12.7, 12.4; UV–vis (CH2Cl2) λ (nm) (ɛ, M−1 cm−1) = 526 (70,000), 497 (20,000), 369 (15,000), 274 (43,000); IR (KBr, cm−1): ν = 3193, 2923, 2853, 1639, 1582, 1532, 1471, 1421, 1385, 1336, 1319, 1212, 1188, 1113, 1072, 1041, 979; FAB+-MS m/z (nature of peak, relative intensity): 1402.1 ([M + H]+, 100); Anal. Calcd for C86H135BF2N4O8: C, 73.68; H, 9.71; N, 4.00. Found: C, 73.41; H, 9.50; N, 3.76.
4.3.2 Compound 12
The compound was prepared from 0.078 g of 3,4,5-tris-dodecyloxy-benzoic acid (0.11 mmol), 0.024 g of 4 (0.057 mmol), 0.044 g of 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (0.23 mmol), 0.035 g of 2,2-dimethylaminopyridine (0.23 mmol) and 20 mL of distilled CH2Cl2 to give 0.062 g of 12, recovered as an orange powder after precipitation (62%). 1H NMR (CDCl3, 300 MHz): δ (ppm) = 7.66 (s, 2H), 7.51 (s, 2H), 7.09 (s, 4H), 4.06–3.99 (m, 12H), 2.52 (s, 6H), 2.33–2.26 (q + s, 4 + 3H, 3J = 7.5 Hz), 1.87–1.70 (m, 12H), 1.57–1.25 (m, 114H), 0.99 (t, 6H, 3J = 7.5 Hz), 0.90–0.85 (m, 18H); 13C NMR (CDCl3, 75 MHz): δ (ppm) = 165.9, 154.0, 153.5, 142.1, 138.6, 137.4, 134.4, 132.9, 130.9, 129.1, 126.5, 122.7, 106.2, 73.8, 69.7, 32.1, 30.5, 29.9, 29.84, 29.78, 29.7, 29.53, 29.51, 26.2, 22.8, 17.2, 14.8, 14.2, 13.3, 12.7, 12.4; UV–vis (CH2Cl2) λ (nm) (ɛ, M−1 cm−1) = 526 (100,000), 493 (sh, 45,000), 360 (30,000), 272 (83,000); IR (KBr, cm−1): ν = 3191, 2917, 1850, 1640, 1581, 1533, 1468, 1418, 1390, 1330, 1319, 1212, 1187, 1112, 1085, 1041, 978; FAB+-MS m/z (nature of peak, relative intensity): 1738.2 ([M + H]+, 100); Anal. Calcd for C110H183BF2N4O8: C, 76.00; H, 10.61; N, 3.22. Found: C, 75.76; H, 10.42; N, 2.98.
4.3.3 Compound 13
The compound was prepared from 0.133 g of 3,4,5-tris-hexadecyloxy-benzoic acid (0.16 mmol), 0.030 g of 4 (0.072 mmol), 0.055 g of 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (0.29 mmol), 0.035 g of 2,2-dimethylaminopyridine (0.29 mmol) and 20 mL of distilled CH2Cl2 to give 0.070 g of 13, recovered as an orange powder after precipitation (47%). 1H NMR (CDCl3, 300 MHz): δ (ppm) = 7.66 (s, 2H), 7.51 (s, 2H), 7.09 (s, 4H), 4.06–3.99 (m, 12H), 2.52 (s, 6H), 2.33–2.29 (q + s, 4 + 3H, 3J = 7.5 Hz), 1.85–1.72 (m, 12H), 1.53–1.25 (m, 162H), 0.99 (t, 6H, 3J = 7.5 Hz), 0.90–0.85 (m, 18H); 13C NMR (CDCl3, 75 MHz): δ (ppm) = 165.9, 154.0, 153.5, 142.1, 138.6, 137.4, 134.4, 132.9, 130.9, 129.1, 126.5, 122.7, 106.2, 73.8, 69.7, 32.1, 30.5, 29.9, 29.81, 29.80, 29.7, 29.6, 29.5, 26.2, 22.8, 17.2, 14.8, 14.2, 13.3, 12.7, 12.4; UV–vis (CH2Cl2) λ (nm) (ɛ, M−1 cm−1) = 526 (100,000), 495 (sh, 46,000), 363 (33,000), 274 (87,000); IR (KBr, cm−1): ν = 2917, 2849, 1641, 1580, 1539, 1466, 1382, 1322, 1276, 1258, 1188, 1114, 979; FAB+-MS m/z (nature of peak, relative intensity): 2075.0 ([M + H]+, 100), 2054.1 ([M − F]+, <10); Anal. Calcd for C134H231BF2N4O8: C, 77.56; H, 11.22; N, 2.70. Found: C, 77.37; H, 10.95; N, 2.35.
Acknowledgments
This work was jointly supported by Louis-Pasteur University through the European School of Polymer and Materials Chemistry (ECPM), the "Centre national de la recherche scientifique" (CNRS) of France and the ANR Contract ANR-05-BLAN-0004-01, FCP-OLEDs. We warmly thank Dr. Pascale Jolinat and Hani Kanaan from the LEGT in Toulouse for performing preliminary OLED devices.