

1 Introduction
The solubility behaviours of a large variety of compounds are of key importance in many natural processes or in fields of activities by humans. For some time, attention has been focused on the solubility of gases such as carbon dioxide or dioxygen in freshwaters as well as in salted ones, e.g. human blood [1,2]. The dissolution and re-precipitation of solid compounds, e.g. calcium carbonate (calcite, aragonite, …), between rocks and water [3,4] or between bones and biological fluids [5–7] are of great concern in geology as well as in pathological sciences. Attention has been recently focused on the physical outcome of cargos accidentally spilled because of shipwreck or other sea event [8–12]. It is, indeed, worth wondering about their fate: indeed, in the event of dissolution of a certain part of a given cargo, which will be this part and in which amount? Is this amount dependent upon temperature and/or water salinity? What is its time evolution?
The literature contains many descriptions of the solubility phenomenon plus others about the effects of either salinity [13–20] (this in a rather phenomenological way) or temperature [1,2,21]. However, to our knowledge, until now no rigorous and synthetic study of the combination of both effects has been reported in the literature. Moreover, most of the people in charge of field measurements have no background in thermodynamics, though they need a precise knowledge of the basic rules that would allow them to correctly use literature data and make the possible, and sometimes necessary, approximations.
Concerning the present and preliminary study, focus will be on only the dissolution/precipitation of solids in more or less salted water and in the simple case where the solid separates pure (no solid solution). Indeed, this simplification of the question under study is not only helpful, but also relevant because it corresponds to many practical cases. The rules governing the behaviour of the dissolution/separation of partially soluble liquid compounds are fundamentally the same, but the formalism is more complicated because the composition of the phase that separates at the solubility limit varies with salinity and/or temperature. This case study will be presented later in another report.
About the restricted case mentioned previously and on condition of knowing the solubility at a single salinity–temperature couple, our aim is to propose a way to calculate the solubility of a solid at any salinity and temperature from some other physical properties of the system. Several possible approximations will be also proposed.
2 Fundamental law of phase equilibria
Let us consider a liquid solvent, A1, and a compound, A2. At a given temperature, the equilibrium between the pure solid state of A2 and its state in solution in A1 can be written as expressed in Eq. (1) [21,2] on condition to consider A1 as a simple liquid solvent or a liquid mixture, e.g. water and salt:
| (1) |
| (2) |
| (3) |
In explicit form, it is written as:
| (4) |
In Eq. (4), the superscript ∗ reads for “pure”; the corresponding free energy then depends on both the temperature and pressure. But since at a pressure close to the atmospheric one, the dependence is weak, it allows one to omit the term, P, which should be enclosed in the brackets. More specifically, this is also consistent with the hypothesis of a constant pressure. About f2, it is the activity coefficient defined with respect to a reference state chosen as the pure component, 2 (x2 = 1); f2 tends to 1 when x2 tends to 1. It is worth noting that, on condition the pressure be close to 1 bar and the reference state be the pure component, the symbol ∗ can be replaced by the symbol of the standard state,0.
Another reference state like, for instance, the ideal solution corresponding to the infinitely dilute solution may be chosen in solution. Then, Eq. (4) becomes:
| (4′) |
But, if the composition variable is molarity, Eq. (1) becomes:
| (5) |
When Eq. (4) is used, the solubility appears as a mole fraction, whereas with Eq. (5) it appears as a molarity. Once obtained, any of these values can be easily converted into the other one.
One should also note that comparing Eqs. (4) and (4′) leads to:
| (6) |
3 Evolution of solubility with salinity, at constant temperature. Role of the Setchenov equation
3.1 Form of the phase diagram
Addition of salt to a three-component mixture (water, solute and salt respectively denoted 1, 2 and 3) at a constant temperature, T, induces changes in the concentrations of all of the three components of the system, x1, x2, and x3, respectively. So, from a general point of view, it is a rule to represent the phase equilibrium line in a triangular diagram such as that in Fig. 1a. The pure solid phase, 2, is in equilibrium with every saturated solutions; so as shown in Fig. 1a, all of the solid–liquid tie lines converge at the point (x2 = 1, x1 = x3 = 0). The dotted area corresponds to the case where, in practice, the system undergoes a maximum salinity value, denoted salmax in this figure: for example, the maximum salinity for the sea-water or biological fluids like blood corresponds to a mole fraction around 0.01. The values in the dotted area may correspond to various situations such as those observed in an estuary. Fig. 1b displays the ordinary representation (solubility–salinity) denoted (sol, sal), which is right according to thermodynamics, but less friendly user for fundamental analyses. However, this kind of drawing is the most common representation of experimental data. In most of the cases, molarities are used instead of mole fractions, but the latter are mentioned in Fig. 1b as possible options in order to facilitate comparisons of Fig. 1a and b. The dotted area on Fig. 1b indicates the area where the salinity can be the highest depending on the specificity of the medium under study (e.g. sea-water). Fig. 1a and b constitutes not only a useful visual summary of the effects by the thermodynamic variables on solubility, but also a reminder of the definitions used in the equations below.
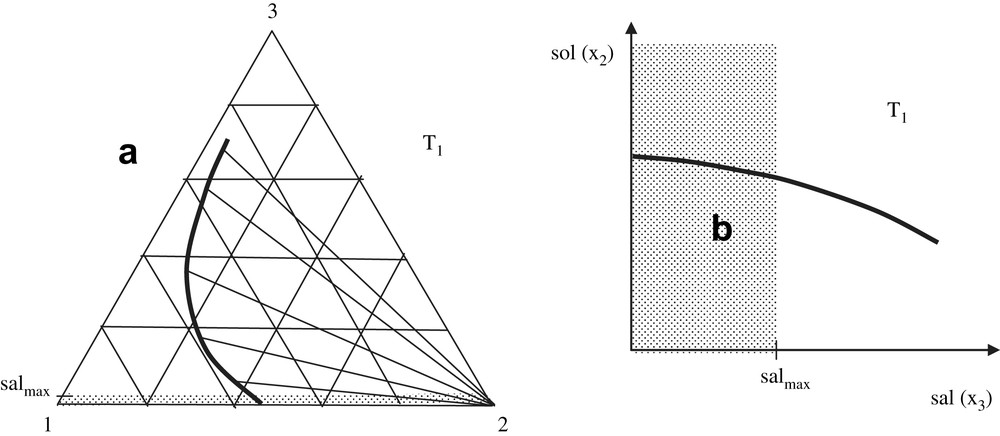
(a) Solubility line at constant temperature for a component 2 separating as pure solid from a mixture with water (component 1) and salt (component 3), in a triangular representation. Tie lines with the pure component 2 are shown as well as the area where salted water may be found in the environment; salmax is the mean salinity of the sea-water as well as of biological tissues such as the blood. The variation of solubility when salinity x3 is raised corresponds to a “salting-out” (it decreases). (b) The same solubility line in an orthogonal representation.
3.2 Equation of the equilibrium line using the mole fraction scale
Equation (4) gives the most straightforward way to get the solubility under the form of a mole fraction:
| (7) |
| (8) |
In pure solvent, Eq. (8) becomes:
| (9) |
Then the ratio between Eqs. (9) and (8) leads to:
| (10) |
The empirical Setchenov equation is written as:
| (11) |
| (11′) |
Equation (10) clearly shows that the solubility ratio directly depends on the activity coefficients, which in turn depend on the concentration of component 2, salinity and temperature. Relation (10) can be expressed in a different way by using the same method as previously from equation (4′) at first for salted solutions and then for solutions in pure solvent. On condition of denoting as Δfusg2θx the standard free energy in the present state of reference, one gets:
| (12) |
| (12′) |
| (13) |
| (14) |
The interest of these relations is that, in dilute solutions, the activity coefficients tend to 1; this is often the case for saturated solutions when the salt is missing. It ensues that γ2x0 ∼ 1; this is why the solubility ratio is often described as the activity coefficient of solute 2 in a saline solution and why the Setchenov relation may be considered as an empirical expression of this activity coefficient.
However, few data about this dependence have been obtained within this scale of compositions (x2), and the use of molarity may appear as offering more user-friendly relationships and corresponds, in fact, to richer series of experimental data [5,8–11]. When some of them are expressed in g L−1, they are easily converted into molarities.
3.3 Equation of the equilibrium line using the molarity scale
Use of Eq. (5) as a basis and application of a treatment similar to the one in Eqs. (12)–(14) lead to the following expression of Setchenov relationship in mol L−1:
| (15) |
In dilute solutions, as these activity coefficients tend to 1, a common approximation is γ2c0 ∼ 1. Then, the solubility ratio gives the activity coefficient in saline solution, and the Setchenov equation provides one with its empirical expression (see Section 3.2).
When the mixture under study consists of electrolytes, it is worth recalling that relation (15) becomes [1]:
| (16) |
3.4 Possible approximations and practical use of these relations
Equations (14) and (15) can be replaced with Eqs. (14′) and (15′). At this stage, Eqs. (11) and (14′) are alike:
| (14′) |
| (15′) |
They show that determining solubility in pure solvent and at several salinity values allows one to determine KzS and then to get, by interpolation, solubility values at other salinities.
Eqs. (14) and (15), by the use of Eq. (10), write as well:
| (14″) |
| (15″) |
On condition the following approximation, γ2x0 ∼ 1 or γ2c0 ∼ 1, be valid and Eq. (5) be taken into account, Eqs. (14) and (15) can be rewritten as:
| (14‴) |
| (15‴) |
These equations show that, when the solubility of the solid in pure solvent is small enough, KzS depends only on the temperature. This result, which was found experimentally in [9], of course depends on the nature of the salt and solute under study [9,15,22,23].
To avoid experiments one may be tempted to use the thermodynamic data reported in the available Tables, which sometimes give also the standard free energy of fusion. Another way is to transform these data from:
| (17) |
| (14″″) |
| (15″″) |
4 Evolution of solubility with temperature, at constant salinity
4.1 Basic treatment
This part of fundamental thermodynamics about the solubility of solid compounds is quite classical; it is particularly simple when none of the concentration variables, e.g. mole fraction and molality, structurally depends on the temperature.
On condition to use the same approximations as in Section 3, namely: (i) P set at a constant value and (ii) the solid phase containing only component 2, and on condition to also set salinity at a constant value while varying the temperature, the variance is again 1. In the phase diagram, the equilibrium is described by a line, and its equation can be found from Eq. (4).
From relation (8) written as follows:
| (18) |
| (19) |
Integrating between temperatures T1 and T2 leads to:
| (20) |
The calculation of solubility data at any temperature, T2, from data at T1 requires to evaluate the activity coefficients from:
| (20′) |
Now, from Eq. (4′) one can write that:
| (21) |
| (22) |
Similarly, from Eq. (5), one gets:
| (23) |
This equation contains some subtle temperature effects issued from the simple effect of temperature on molarity, i.e. with no change in the solution content.
However, in short ranges of temperature, one can write
| (24) |
4.2 Empirical treatments and common approximations
When the interval of temperatures between T1 and T2, (T2 − T1), is small enough, a common approximation is to assume that Δfush2∗ is constant in Eq. (20), which allows one to express the integral on the right as:
| (25) |
It is similar for Eq. (22):
| (26) |
| (27) |
According to experiments [16,9,1,2], when the Setchenov constant [Eqs. (14), (15), (14′), (15′), (14”), (15”)], is independent of concentrations, it is almost unaffected by temperature; this seems particularly true with metallic chlorides and between 25 and 40 °C [15,19]. Indeed the rare measurements of a temperature effect on the Setchenov coefficient have demonstrated a slight effect, but as they had been performed in the molarity scale, the observed effect may have been induced by the temperature-dependency of the molarity. It is true that, in a mixture, a massive presence of salts tends to dominate the interactions. So, it would be worth considering to what extent salt activity is temperature-dependent, since, at each step of temperature, it affects the activities of the other components.
It ensues that a possible approximation is:
| (28) |
When the Setchenov approximation and Eq. (28) are both valid, salinity has only a slight effect upon the way the solubility of any compound is affected by temperature. Another way to reach the result is the following.
According to Eq. (11), when the salinity is weak and the solutions are dilute, , and then the use of Eq. (22) leads to:
| (29) |
Relation (29) means that the solubility varies with temperature and at constant salinity, “sal” = constant, it varies as in the absence of salt (“sal = 0”).
5 A proposal to evaluate solubility from data obtained at other salinity and temperature values
Fig. 2 exhibits the way one can pass from a solubility value in the state 1 (sal1, T1) of the system to the solubility value in the state 2 (sal2, T2). The large-dashed line shows how one can, at first, follow the coexistence line at T1 until reaching (state 1′).

Solubility surface obtained from figures such as Fig. 1a when the temperature is varied. The triangle of Fig. 1a is used at three temperatures; the solubility is assumed to be enhanced by the rise of temperature. A dashed line from point 1 to point 2 shows how a coexistence line, at constant temperature, can be followed from salinity sal1 to salinity sal2 (also called in the text), point 1′, then at constant salinity, by varying the temperature from the point 1′ (at T1) to the point 2 (at T2). Point 1 could have been chosen at salinity 0. A second path (dotted line) shows how, in a medium where salinity is spontaneously limited (e.g. sea-water), one can determine the solubility from a value at another salinity, even null.
The equation representative of the whole path can be written in either a rigorous way or in a simplified formulation as shown below.
5.1 Rigorous equation
The evolution of the solubility of component 2, at first along the path from state 1 to state 1′, is then described in the rigorous form by Eq. (10) or (11) in the symmetrical system of reference, or by Eq. (15) in the unsymmetrical system; one should note that Eqs. (11) and (15) include Setchenov's formula. Then, the changes in solubility between state 1′ and state 2 are described by Eq. (20) or (22) in the symmetrical system or the corresponding ones in the unsymmetrical system.
By using Eq. (10) to pass from state 1 (T1, x21, sal1) to state 1′ (T1, , ), i.e. along an isotherm, then by using Eq. (20) along an “iso-salinity” path to go from state 1′ (T1, , ) to state 2 (T2, x22, ) one gets:
| (30) |
Both f2 parameters can be estimated from Eq. (11′) written at T1 and at T2, respectively. So the ratio of activity coefficients in Eq. (30) writes:
| (31) |
Combining Eqs. (30) and (31) leads to a rigorous equation expressed as:
| (32) |
By using the unsymmetrical reference system, this equation becomes:
| (33) |
On condition of admitting the Setchenov relationship, Eq. (33) rigorously describes the evolution of the solubility limit of a pure solid further to changes in temperature and salinity. In principle, this equation can be used in both senses: the known terms are either x21, x22 and Δfush2θx plus, for example, KxS, (left member of Eq. (33)), and then one gets information about γ20, or those on the right, and then the x22 value can be calculated from x21 one. This second case is the one met in accidental environmental events when practical solutions have to be found from partial data or had been previously included in codes written for decisional tools; then these codes must contain a sufficient amount of data to allow calculation. There is no doubt that getting the detailed knowledge of the physical characteristics (as contained in Eq. (33)) of any chemical liable to be implicated in accidental events remains impossible. So, it is worth using simplified relationships, such as those presented in the next paragraph, while being aware of their limitations deduced from comparison with Eq. (33).
One should, however, note that models able to calculate activity coefficients are available [1,2,26] and could be used in Eq. (33). Unfortunately, calculations are too time-consuming whenever measures have to be quickly taken because of an accidental event.
5.2 Dilute system, simplified Setchenov's formulation
In the absence of salt, when the solubility is low at both temperatures, γ20 at both temperatures can be approximated to 1. If, in addition, KxS is admitted to be temperature-independent, KxS (T2) ∼ KxS (T1) = KxS
If Δfush2θx does not depend on temperature or if (T2 − T1) is small, the integral can be also approximated, which leads to the following approximation of Eq. (33):
| (34) |
If the start of experiments, or calculation, is at null salinity, then xsal = 0 and:
| (35) |
Expressions (34) and (35) are far simpler than Eq. (33), and everyone knows exactly which approximations have been made to get them and, thus, when they have to be used in a more elaborated form.
In many cases, particularly when slightly soluble compounds are dissolving in the marine environment, the mole numbers contained in 1 L of mixture are clearly smaller for solutes than for water: n2, n3 ≪ n1 ∼ (m1/M1) = 1000/18 = 55.5, and thus, n2 ∼ c2 and n3 ∼ c3, which leads to rewriting Eq. (34) as:
| (36) |
Let us examine the Setchenov equation in molarity and mole fraction scales; respectively, it gives:
| (37) |
| (38) |
| (39) |
Then Eq. (36) becomes:
| (40) |
It is worth noting that this expression uses molarities in each term except the enthalpy of fusion, which results from a rigorous analysis of temperature effects made impossible when direct molarities are used in the calculations.
Obviously, when salt effect is used in extraction processes, the salt concentrations are far too high to allow the use of these approximations. But, in the case of an accidental marine pollution by chemicals, Eq. (35), and above all Eq. (40), can provide one with a correct evaluation of the solubility under marine conditions at the very temperature of sea-water despite the dependency of this temperature on weather conditions and the period of the day (daylight against night). These formulae are worth using because the amount of data to be known is small: only are required the melting enthalpy at P = 1 bar, the melting temperature, Tf, and the Setchenov constant, KxS or KcS. The two first parameters are available in the Tables of thermodynamics, and if the Setchenov constants have not been measured, they can be calculated by incremental methods [10,16,20,22].
6 Conclusion
By using the fundamentals of thermodynamics about phase equilibria and the effect of salinity on the solubility limit of any solute proposed by Setchenov we established a relationship between the solubility at a first state of salinity and temperature (sal1 and T1) and the one in a second state (sal2 and T2). This relation may use mole fractions in the symmetrical system of reference (Eq. (32)) or in the unsymmetrical one (Eq. (33)). In dilute solutions including mixtures in sea-water, and on condition the variation of temperatures be small, these equations can be simplified (relations (35) and (36), respectively) in order to foresee realistic solubility values from only a set of essential parameters: the melting enthalpy and the melting temperature of the pure solute under study, the Setchenov coefficient. A less rigorous equation, namely Eq. (40), relies on the use of molarities. Among these essential parameters the two first one have to be found in Tables, whereas the third one has to be measured or estimated from increments found in literature. Some care has, however, to be taken with respect to the conversion of data from a system of concentrations to another one since molarities are more difficult to use in studies where temperature varies. Similarly, the reference systems for activity coefficients have to be taken into account. Then, the use of Henry's constants plays a fundamental role. When chemicals are characterised by unusual values of thermodynamic parameters, particularly high solubility values, coming back to the more elaborated relations in order to study specific treatments of the issue of concern is a must. The formalism used in the present study cannot be directly applied to any kind of solubility limit: in particular, it is inapplicable for two reasons when the pure form of the solute under study is liquid. Indeed, i) the phase that separates when the solution is saturated is a mixture, and ii) the parameter equivalent to the enthalpy of fusion becomes a transfer coefficient, and then it is less easily handled than the previous enthalpy.
Appendix Chemical potentials in several reference systems
The chemical potential, μ, of any compound is an intensive variable characteristic of the compound defined as “the partial Gibbs free energy” of the compound. If the compound is pure, it is simply the molar free energy. The expression of the relationship between a chemical potential and the other state variables, e.g. pressure, temperature, composition, is let to the free choice of the scientists working about a given system.
For a pure perfect gas at the temperature and pressure, T and P, respectively, one demonstrates that:
| (A1) |
In a mixture, let us denote this gas as 2, then the chemical potential is written as:
| (A2) |
The expression of μ2 in a liquid mixture can be given on starting from the equilibrium conditions with respect to diffusion between the liquid mixture and its vapour, namely for component 2:
| (A3) |
| (A4) |
Quite formally, let us consider the liquid/vapour equilibrium for the pure component 2 whose vapour pressure is called P2∗, then, Eq. (A3) can be rewritten as follows where the chemical potentials are equal to molar Gibbs free energies:
| (A5) |
Combining Eqs. (A5) and (A4) leads to:
| (A6) |
| (A7) |
Fig. A1, which displays the quite general behaviour of partial vapour pressures above a mixture of two liquids totally miscible, shows that when x2 is close to 1, i.e. close to the pure component 2, p2 linearly varies along a straight line starting from P2∗ at x2 = 1 (pure component 2) and ending at 0 for x2 = 0 (pure component 1). This line is named the Raoult line for component 2, and the mixture behaviour is called “ideal”. Along this line, one gets: Variations of the partial pressures of two totally miscible components 1 and 2, respectively denoted p1 and p2, at a given temperature. The total vapour pressure is P = p1 + p2 where p1∗ and p2∗ are the vapour pressures of components 1 and 2, respectively. The pressures are represented versus the mole fraction of component 2 in the liquid mixture. In dilute solutions (small x2), p1 and p2 both vary linearly with x2; the straight line corresponding to p1 varies between p1∗ and zero (Raoult's law), p2 varies between zero and p2θx (Henry's law).
By using this relation in equation (A6) one gets the expression of the chemical potential as a function of the liquid mixture parameters in ideal solutions and with a pure component 2 set as reference:
| (A8) |
When x2 values are clearly less than 1, the p2 curve goes away from the Raoult line, and then the chemical potentials are usually written as:
| (A9) |
Instead of using the behaviour close to the pure component 2, one can use another linear behaviour of p2(x2ℓ), namely the one in very dilute solutions, x2ℓ → 0, where, as shown by Fig. A1, one gets:
| (A10) |
One should note that this straight line is called Henry's straight line, H2x being the Henry constant of component 2 in the mixture where the molar fraction is the composition variable.
By combining Eqs. (A10) and (A6), one gets another expression of μ2 (which obviously keeps the same value when T and x2 stay unchanged):
| (A11) |
| (A12) |
The parameter:
| (A13) |
| (A14) |
When x2 values move clearly away from zero, the p2 curve goes away from the Henry line, and then the chemical potentials are usually expressed as:
| (A15) |
Another possible choice is to always use Henry's line on condition the molarity, c2, be the concentration variable.
| (A16) |
In the case of two components, one gets:
| (A17) |
| (A19) |
| (A20) |
In a system where water is the solvent, the mixing of Eqs. (A6) and (A20) leads to:
| (A21) |
According to Eq. (A21), g2θcℓ(T) is defined as:
| (A22) |
Finally, out of the zone of linear variation of p2 with x2, the chemical potential is usually expressed as:
| (A23) |
In order Eq. (A21) remains valid over this range of concentrations, the limit condition, γ2c → 1 as x2ℓ → 0, has to be added to the definition of γ2c given by Eq. (A23).
In the use of tables of data, careful attention has to be paid to the reference states to which the data under study are related. The conversion of all data in a same system is required to comply with consistent calculations; it mainly relies on the use of Eqs. (A14) and (A22), or maybe (A19), for the conversion of the free energy of phase changes. Equations (A9), (A15) and (A23) all give the expressions of chemical potentials in several cases.
From these equations, on condition to combine μ2ℓ and μ2c for example along a fusion line, one gets successively:
| (A9′) |
| (A15′) |
| (A23′) |
But, by using Eqs. (A14) and (A15) can also be written as:
| (A15″) |
Similarly, when water is the solvent, the use of Eq. (A22) allows one to write Eq. (A23′) as follows:
| (A23″) |
Comparison between Eqs. (A15′) and (A15″) on the one hand, and Eqs. (A23′) and (A23″) on the other hand respectively leads to:
| (A16‴) |
| (A23‴) |
Equations (A16‴) and (A23‴) allow the conversion sometimes necessary between free energies of fusion measured under different conditions.