

1 Introduction
The study of molecular- and nanophosphors [1], that is luminescent dyes, lanthanide complexes, transition metal clusters and inorganic nanocrystals, is largely motivated by the prospect of original specific applications such as electroluminescent devices [2], integrated optics [3] or biological labels [4].
Concerning biological applications, fluorescent organic compounds have been extensively used for the visualization of the different components of biological systems [5]. An interesting issue is to track labeled individual species during in vitro or in vivo experiments, but the use of organic dyes is drastically limited for that purpose because of their rapid photobleaching. A few years ago, the use of isolated inorganic nanocrystals such as semiconductor quantum dots (QDs) was proposed since they exhibit a bright and almost stable luminescence permitting single particle observation over a long period of time as compared to current organic fluorophores [6]. Many works done in this field have shown that these inorganic particles, specifically conjugated to biological macromolecules, provide a new tool for biologists to study the individual action of biologically active species [7].
For applications in solid-state dye lasers, electroluminescent devices and integrated optics, molecular- and nanophosphors have to be assembled in transparent composite materials. During the last 20 years, numerous siloxane-based hybrid organic–inorganic materials exhibiting light emission properties were generally obtained by embedding or grafting organic chromophores in sol–gel derived hybrid matrices [8]. For example, works made in our group [9] and in others [10] concerning solid state dye lasers have shown that the prospects for these hybrid materials are extremely promising as hybrid dye lasers exhibit low threshold powers for laser action (a few millijoules), long laser lifetimes (106 pulses), and operation at high repetition rates (10 Hz). However, as for biological labels, the applications for dye-doped systems are still to some degree limited by the progressive photo- and thermobleaching of the dyes and the development of inorganic new phosphors is clearly necessary to emerge beyond the demonstration level.
This short review focuses on three areas of our work on inorganic nanophosphors:
- • the sol–gel assembly of metal chalcogenide QDs which has been initiated in our group and recently developed by S.L. Brock and co-workers;
- • the synthesis and the biological functionalization of lanthanide doped-oxide nanoparticles (NPs) exhibiting bulk behaviour for their luminescence properties;
- • the synthesis of luminescent copper iodide clusters and their integration in siloxane-based films.
2 Sol–gel assembly of metal chalcogenide QDs
Since the beginning of the 1980s, impressive amount of work have been done in the field of semiconductor QDs, especially on chalcogenide compounds [11]. Main issues concerned their controlled synthesis for the understanding and optimization of new physical properties. These systems indeed exhibit the so-called quantum confinement effect that is one of the most impressive size effect observed in nanomaterials. At the beginning of the 1990s, improvement of the elaboration processes allowed the optimization of the emission properties of these particles, so that emission efficiencies could almost compete with common luminescent dyes but with an improved photostability [12]. This opened the way to intensive researches on the applications of these systems, mostly as light emitting materials and new probes for biological labeling.
Different materials (bulk or thin films) can be made using NPs as one of their constituent. A simple and easily scalable process is the elaboration of composite materials in which particles are dispersed within a dielectric host matrix (polymer, sol–gel silica). Another issue is the elaboration of materials using the NPs as building blocks. This allows the optimization of their volume fraction, may preserve the chemical access to their surface and control interparticle distances. The latter point finds its interest in original properties related to interparticle interactions that are distance dependant, so that the elaboration of 2D or 3D networks of particles with controlled structures and lattice parameters is a subject of intense activity [13].
The colloidal processing of ceramics through random aggregation of NPs is an active field of researches in materials sciences [14]. The controlled aggregation of highly concentrated colloids provides the opportunity to reliably produce ceramic films and bulk forms through much easier scalable processes than organized networks, still allowing a control of micro- or nanostructure. In this field, the elaboration of optically transparent materials represents the ultimate challenge. A transparent material rather than a diffusive powder will indeed be obtained only if the aggregation is well controlled so that the structure of the aggregates preserves the homogeneity of the material in the visible wavelength scale.
The sol–gel process relies on the controlled formation of such lacunar aggregates within the solution, which is called a “sol” [15]. Percolation of the aggregates to obtain a solid structure is achieved either from rapid solvent evaporation (deposition of thin films by spin or dip-coating) or simply by their continuous growth within the sol. This moment corresponds to the sol–gel transition, a gel being a rigid skeleton of particles enclosing a continuous liquid phase. The careful removal of the solvent from the liquid phase yields the final solid material. This material can be an aerogel with a very low density if the drying is achieved under supercritical conditions or a dense xerogel if the drying is made slowly at low temperature (20–100 °C) to avoid fractures from capillary stresses [15].
Until recently, sol–gel processing of transparent nanoparticulate materials was restricted to a limited number of compounds, mostly oxides and especially silica. The main barrier for the extension to other compounds relies on the difficulty of the synthesis of highly concentrated colloids with a controllable state of dispersion. We discuss here on an original process that was successfully applied to chalcogenide QDs, allowing the elaboration of xerogels (thin films or bulk) or aerogels with interesting optical properties.
2.1 Thiol stabilized concentrated solution
The beginning of this work is the experimental observation that surface derivatization of CdS NPs with 4-fluorophenylthiol allows their stabilisation in common organic polar solvent with an extremely high concentration [16]. This stabilization ability is much more important than it is for other complexing molecules which have been previously used and, more particularly, as compared to the nonfluorinated phenylthiol. The latter had been shown previously to be an excellent stabilizer for the NPs in pyridine, but the use of the fluorinated phenylthiol was found to induce an extraordinary improvement of the dispersion ability of the particles in polar organic compounds. Nuclear magnetic resonance (NMR) studies of the surface structure revealed that it is almost the same with or without the fluoride substituent. The difference of stability was thus attributed to the large dipole moment of the fluorophenylthiol, allowing a strong solvatation by polar organic solvent molecules. The resulting thick structured solvent shell around the particles prevents their aggregation and thus drastically improves their stability. Concentrations of more than 30% net weight have thus been obtained in common polar organic solvents such as acetone or tetrahydrofuran.
2.2 Sol–gel transition in CdS colloids
It is well known that thiol can be easily oxidized under soft conditions into disulfides. In the case of thiol stabilized particles in non complexing solvents, such an oxidation leads to a progressive random aggregation of the particles following the scheme described on Fig. 1. It was found that controlled oxidation of the stabilizing thiols could be achieved simply by the addition of H2O2 [17]. The time evolution of a CdS sol depends on the H2O2 to grafted thiolate molar ratio. Addition of a small amount of H2O2 does not induce any visual evolution of the solution until a minimum (about 0.2 for 0.1 mol/L in CdS) is reached for which gelation is observed. At this point, fractal aggregates that have progressively grown within the solution percolate so that a solid network of NP completely fills the solution. In the case of concentrated sols, no visible light diffusion is observed while the sol turns into a gel, showing that the fractal aggregates do not have any characteristic structural dimensions in the order of the visible light wavelength.
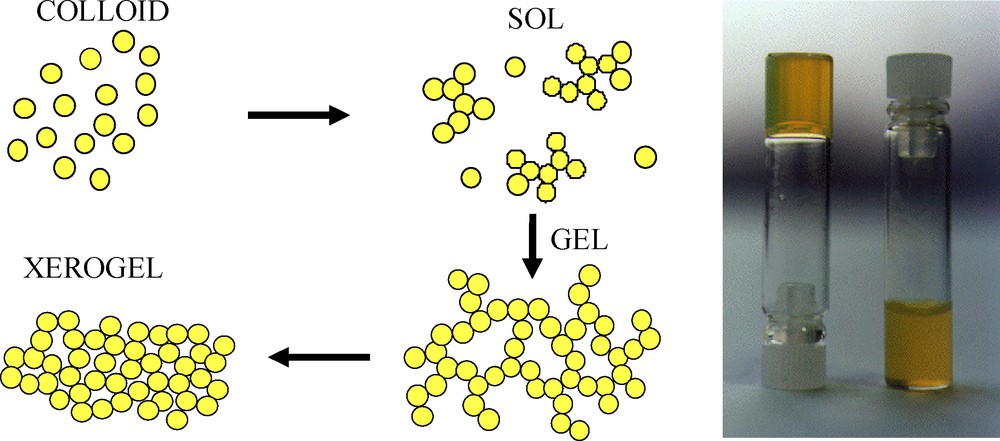
Sol–gel aggregation of CdS nanoparticles (NPs) through their controlled aggregation induced by the progressive removal of surface thiol group (left) and picture of a gelified CdS colloid (right).
2.3 Nanoparticulate materials: xerogels (bulk or thin films) and aerogels
Observation of sol–gel transition in CdS colloids opens the way toward the elaboration of transparent solid nanoparticulate materials. These materials are of interest due to their potentially interesting optical and electrical properties, resulting from a preserved nanostructure and a direct contact between the particles. Sol–gel nanoparticulate materials, obtained after removal of the solvent from a randomly aggregated assembly of particles, are mainly of three different kinds:
- • xerogel thin films are obtained by deposition onto a substrate of a sol of partially aggregated NPs. These materials are the most common application of the sol–gel process and have been successfully obtained in the case of CdS sols [18]. The obtained transparent films exhibit the same optical properties as the initial particles for thermal treatments less than 150 °C, after which removal of the capping molecules by thiolysis induce the sintering of the particles;
- • xerogel monoliths may be obtained after careful drying of a gel, but as commonly observed in such processing, capillary stresses induced by solvent evaporation is a strong limitation to obtain bulk materials without cracks. Such stresses may be avoided by performing the removal of the solvent through supercritical CO2 drying. The obtained materials are highly porous, since the structural integrity of the framework is mainly maintained through the supercritical treatment;
- • the elaboration of chalcogenide areogel through direct application of our gelation process was demonstrated [19] and extended to other compounds (GeS2 [20], CdSe/ZnS [21]) by Brock et al. This opens the way toward the investigation of the physical properties of many new systems that exhibit original microstructures in the nanometer size range.
3 Lanthanide-doped oxide NPs
The research on QDs has clearly shown that the colloidal synthesis only constitutes a first step towards the preparation of a nanophosphor suitable for applications. The control of the surface chemistry is also essential both to optimize the luminescence properties and to allow solubilization and functionalization of the particles in the application medium. If complex architectures have been already realized for QD systems, leading to specific chemical or biological interactions, equivalents were rarely reported for lanthanide-doped oxide nanocrystals. We review current approach to the lanthanide-doped lanthanum phosphate and yttrium vanadate nanophosphors, which appear to be among the most promising systems for future applications.
3.1 Colloidal synthesis
The liquid-phase synthesis in high-boiling-point coordinating solvents has been largely developed for processing well-known semiconducting luminescent nanocrystals. The synthesis is generally performed at 200 to 300 °C, leading to semiconductor particles with a good crystallinity and a good control of their size in the 2 to 10 nm range. Numerous examples follow this approach, such as the synthesis of CdSe QDs in trioctylphosphine oxide (TOPO) [13,22], InAs in trioctylphosphine (TOP) [23], ZnSe NPs in hexadecylamine [24].
Lanthanide-doped insulator oxides constitute an important other class of nanophosphors for which different types of liquid phase-syntheses have led to the production of colloidal suspensions [25]. These compounds are well known as commercial bulk phosphors, but their high temperatures of elaboration are difficult to reach in liquid media. However, our group developed the synthesis of lanthanide vanadate (YVO4:Ln) and phosphate (LaPO4:Ln) NPs as well-dispersed and highly concentrated colloids by a simple aqueous route using competition between precipitation and complexation reactions [26]. A core-shell strategy was also reported to improve the chemical stability and the brightness of the colloidal particles, using the growth of LaPO4·xH2O and SiO2 protective layers [27]. Under UV excitation, the three primary colors are furnished by YVO4:Eu, LaPO4:Ce·0.7 H2O and LaPO4:CeTb·0.7 H2O aqueous colloids, which emit red, blue-violet and green light respectively under UV excitation. It is thus possible to screen a large wavelength range for the light emission by changing the nature of the lanthanide-doping ion in yttrium vanadate and lanthanum phosphate matrices (Fig. 2) [28].

Luminescent colloidal suspensions under UV excitation prepared from the three primary colors furnished by YVO4:Eu, LaPO4:Ce·0.7 H2O and LaPO4:CeTb·0.7 H2O aqueous colloids which emit red, blue-violet and green light respectively under UV excitation.
3.2 Luminescence properties of crude NPs
The spectroscopic features of the particle emission are roughly similar to the related bulk materials, that is very narrow emission lines with a millisecond lifetime of the excited states. The luminescence of both YVO4:Eu red and LaPO4:Ce,Tb green bulk phosphors can be described from the ATE mechanism with absorption, transfer and emission steps. First, the two systems present intense and large absorption bands under UV excitation, respectively corresponding to a charge transfer band into the VO43− group and to the 4f-4f5d absorption bands of the Ce3+ in the phosphate matrix. The second step consists in energy transfers: between vanadate groups and to europium ions for YVO4:Eu [29], between cerium ions and to terbium ions for LaPO4:Ce,Tb [30]. For the red phosphor, the emission spectrum is then dominated by the 5D0–7F2,4 forced electric-dipole transitions for which the high intensity is a consequence of the absence of inversion symmetry at the Eu3+ lattice site (D2d symmetry) [31]. For the green phosphor, the emission spectrum displays the typical line spectrum of the terbium 5D4–7FJ transitions between 450 and 700 nm [32]. A large emission band (at 350 nm for the bulk monazite) is also observed due to cerium ions whose luminescence is not completely quenched.
A first important problem to use these aqueous colloids is that the luminescence yields (for example 20% for the red YVO4:Eu [26b]) are significantly lower than for corresponding bulk materials (between 70–90%) and the optimum concentrations of doping-lanthanide ions are higher.
The surface of small particles, where the coordination of atoms differs from the bulk and where different chemical species may be adsorbed, is suspected to be the most important source of alteration of the luminescence efficiency. For aqueous colloids, it can be expected that most of the surface of particles will be covered by OH groups, which are well known to quench the luminescence of lanthanide ions. This effect has been evidenced by transferring YVO4:Eu nanocrystals into D2O where a clear increase of the luminescence yield (from 20–40% for Eu20%) has been observed, confirming that surface OH groups are efficient quenchers of the Eu3+ excited states in red nanophosphors [33].
For YVO4:Eu nanocrystals, a major difference between the emission behavior of the colloid and the bulk material is observed when plotting the luminescence yield as a function of the europium content. In the bulk material, a clear optimum of the luminescence yield (70%) is obtained for europium concentration of 5% [32a]. In the case of colloidal particles, this optimum is no more well defined and occurs in the 15 to 30% europium content, while the maximum efficiency does not go beyond 20%. The reasons why the evolution of the emission yield as a function of the europium content markedly differs from the bulk materials has to be found in the efficiency of VO4–VO4 energy transfers. It is indeed well known that the optimum of the Eu3+ concentration results from two competitive effects: on the one hand, an increase of the europium concentration improves the probability of the energy transfer to europium ions, that is the number of excited centers and thus radiative recombination. On the other hand, there is also an increase of the probability of energy transfer between Eu3+ ions, which increases the efficiency of the excitation capture by non-radiative recombination centres (concentration quenching). The observation of a high luminescence yield for a quite small europium content in the case of YVO4:Eu bulk compounds is related to the very efficient energy transfer through adjacent vanadate species, until the excitation reaches a europium ion. The smooth optimum at 20% observed for YVO4:Eu red nanophosphors is then a clear indication of the alteration of these energy transfers. The main reason for the alteration may be found in the low temperature synthesis, which leads to porous polycrystalline aggregates where structural defects enhance non-radiative processes leading to the desexcitation of the excited vanadate species.
3.3 High temperature strategy to improve luminescence properties
We have recently reported an original process permitting thermal annealing of oxide NPs at high temperature (up to 1000 °C) without a significant growth and without particles adhering to each other [34]. In comparison with usual techniques of preparation of NPs performed at low temperature (from room temperature [RT] to 300 °C), this high temperature technique is able to produce perfectly crystallized particles. Moreover, after thermal treatment, the process allows dispersing annealed particles in water, leading to concentrated aqueous colloidal dispersions of multicomponent oxides containing isolated highly-crystalline particles.
Concerning YVO4:Eu particles, a concentrated aqueous dispersion of crude NPs was prepared early using colloidal techniques such as a precipitation reaction at RT from precursor salts in aqueous solution. The initial colloidal suspension consists in dispersed polycrystalline particles exhibiting a prolate ellipsoid shape with average axial dimensions of 20 and 39 nm. Using the sol–gel technique, crude NPs were then mixed in a silica sol using a copolymer both as a dispersing agent to prevent particle aggregation and as a structure-directing agent to obtain a mesostructured silica network. After silica gelation and copolymer calcination, this leads to homogeneous concentrated dispersion of the crude particles in a highly porous silica matrix (porous volume fraction up to 40%), which ensures the role of a refractory solid dispersing agent. In fact, no significant interaction was detected between the silica matrix and the starting NPs after thermal treatments up to 1000 °C. Besides, in presence of NPs and after the thermal treatment, the porous silica matrix was still amorphous and was not significantly sintered, making possible its chemical dissolution by hydrofluoric acid 2% in excess, without significant alteration of NPs. Finally, the annealed NPs were purified and dispersed in water by sonification using a stabilizing agent. Structural characterizations from XRD and TEM experiments have clearly shown that the thermal annealing turns polycrystalline and highly constrain crude units into YVO4:Eu single nanocrystals without structural transition, coalescence and significant size increase (Fig. 3a, b).
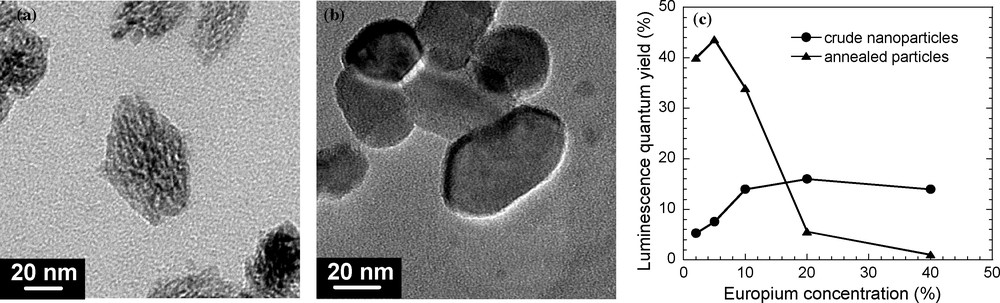
(a and b): TEM images of YVO4:Eu (10% Eu3+) NPs after annealing at 800 and 1000 °C, respectively. (c): Evolution of the luminescence quantum yield as a function of the europium content for crude and annealed YVO4:Eu NPs at 1000 °C.
A first expected interest for this process concerns the impact on the luminescence properties. As previously discussed, the red luminescence properties of YVO4:Eu compounds are governed by energy transfers occurring after the absorption of light, taking place through exchange (VO4–Eu) [35] and multipolar (Eu–Eu) interactions [36]. These transfers are all the more favoured when the wave function overlaps are efficient as in the bulk material, making them highly sensible to structural defects. Under excitation at 280 nm in the orthovanadate band and, in comparison with Y1-xEuxVO4 crude particles, the quantum yield is largely increased in annealed particles (37% instead of 14% for 10% Eu) and the quantum yield of annealed NPs is not significantly changed when the colloidal solution is transferred into deuterated water. The reduction of surface quenching effects in annealed NPs is probably related to the structural reconstruction of NPs at high temperature by the self-sintering process, which decreases the OH adsorption. In fact, the surface area of a powder of annealed particles is only of 10 m2/g, instead of 200 m2/g for crude NPs. This shows a significant decrease of the porosity and of the surface roughness of NPs, leading therefore to a reduction of the number of accessible Eu ions for the adsorption of OH groups.
In addition, while no well-defined maximum is observed on the curve showing the europium concentration dependence of the luminescence yield for crude NPs, it can be noted that the annealed NPs present, as for the bulk material, an optimum of the europium concentration around 5% (Fig. 3c). All these results clearly demonstrate that the energy transfer efficiency is high for annealed well-crystallized NPs, similarly to bulk materials. Therefore, the annealing process furnishes highly bright nanophosphors, which are potential candidates as biological luminescent labels or for integration in transparent luminescent coatings.
3.4 Organic functionalization towards application as biological probes
Another well-known difficulty to use luminescent NPs in different applications concerns the surface functionalization. This is a key step toward applications as biological luminescent labels or to elaborate transparent optical materials since it determines the control of the coupling between the particles and the chemical or biological species of interest.
The surface functionality can be introduced by the following ways, previously developed for QDs:
- • the growth of a silica-type thin layer by condensation of F-R-Si(OR)3 organosilane precursors having the desired F active group;
- • the direct grafting of C-R-F organic molecules where C is a complexing function (phosphonate, thiol, carboxylate);
- • the capping with various polymers (PMMA, silicates).
In the case of aqueous colloids, the first approach which relies on the encapsulation of the particles with a thin layer of functional polysiloxane resulting from the silane condensation has been already studied in many colloidal systems (such as silica [37], boehmite [38], maghemite [39], QDs [40] and lanthanide-doped particles [41]). Silane coupling agents present the advantage to provide a covalent grafting of reactive functions with a high surface coverage and benefit from the versatility of silane chemistry. The bonding of silane molecules at the surface of particles generally requires a preliminary activation step such as the adsorption of silicate ions that will act as a primary layer for the further surface polymerization of the silanes [38]. However, it can be noted that the efficiency of this functionalization process was generally shown indirectly through the modifications of the particle interactions with their environment, without a quantitative characterization of the surface chemistry modifications.
We have recently described the application of this functionalization strategy, using amino-and epoxysilanes in the case of luminescent YVO4 NPs that were shown to be attractive new biological labels [42]. The amino or epoxy functions born by the silane are commonly used for the functionalization of substrates such as biochips for further coupling reactions with bioorganic species. In our study, a special attention was paid to the careful characterization of each step of the functionalization process, especially concerning the average number of organic functions that are available for the final coupling of particles with proteins. This was achieved using conventional analytical techniques (chemical analysis, TGA, NMR, IR spectroscopy). The surface density of epoxy and amino functions was found to be 0.4 and 1.9 functions per square nanometres for silanized particles, respectively.
First results concerning grafting of biological species on YVO4:Eu NPs were obtained using epoxy-functionalized particles for the biological labeling of sodium channels in living cells [43]. In this case, guanidinium groups were covalently reacted with the dangling epoxy functions at the surface of particles. The guanidinium group is the active part of two naturally occurring toxins, tetrodotoxin and saxitoxin which selectively and potently block voltage-dependent sodium channels by plugging the channel's mouth [44]. The efficiency of the functionalization was mainly attested by the physiological properties of the particles that behaved like artificial toxins targeting and inhibiting the sodium channels in a similar way as the natural toxins [43].
The coupling of proteins onto amino-functionalized particles was quantified in the case of α-bungarotoxin that was previously labeled with an Alexa fluorescent dye [45]. This allowed testing the coupling of this protein with amino-functionalized NPs through the use of a homobifunctional crosslinker, namely BS3 which can react in a first step with amino groups at the surface of particles and, in a second step, with amino groups on the protein surface through two succinimidyl ester groups (Fig. 4). The use of a protein tagged with a fluorescent dye has allowed the accurate determination of the number of grafted proteins per particle showing that upto eight proteins were coupled on average. Moreover, in order to use NPs for applications as single-molecule labels, the exact number of proteins per particle at the single particle level has been determined using the stepwise photobleaching of the Alexa dye molecule. Current development concerns applications of these particles to address biological issues such as real-time tracking of single biomolecules labeled by YVO4:Eu NPs.
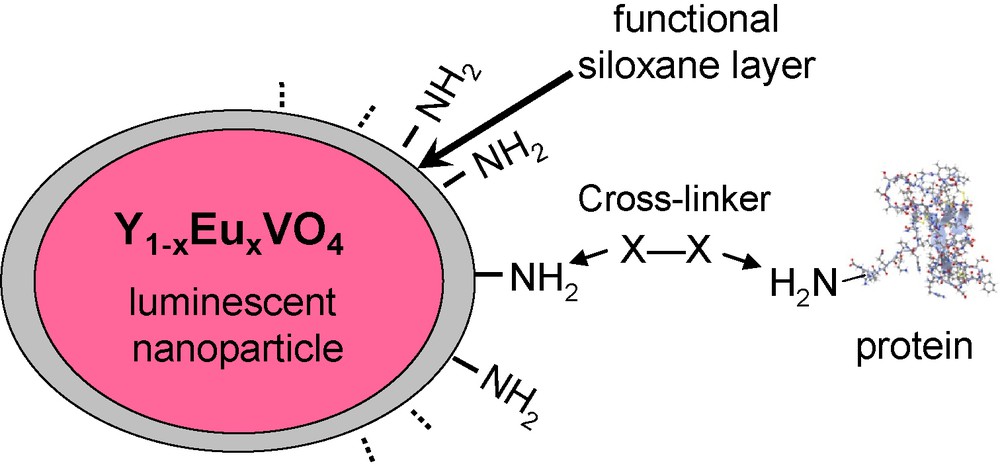
Schematic representation of the coupling of proteins to luminescent nanoparticles (NPs) using a bifunctional crosslinker (X–X).
4 Luminescent molecular clusters in silica-type matrices
Luminescent transition metal clusters represent another class of phosphors to take into account for the synthesis of light-emitting materials (silica-based materials or not). Luminescent inorganic molecular clusters can be considered as promising photoactive species since they usually combine the inorganic structure of the mineral NPs with the monodispersity and the relative easy functionalization of the organic molecules. Their incorporation within sol–gel matrices can thus lead to novel light emitting materials exhibiting both the transparency and processibility properties of the matrix associated with the unique luminescent properties of the clusters. According to their wide range properties, original multifunctional materials can be also expected. In this prospect, several studies have been reported involving the incorporation of transition metal clusters within silica sol–gel matrices. To our knowledge, three family of transition metal clusters have been studied namely, lanthanide-doped polyoxometalates (POMs), molybdenum halogenide and copper iodide clusters. Results concerning these composite cluster-silica materials are presented here.
4.1 Lanthanide-doped POMs
POMs are early transition metal oxide cluster anions formed by condensation of MO6 octahedra. Considering their structures, sizes and properties, POMs are intermediates between small molecules and bulk oxides. They exhibit various topologies and diverse chemical and electronic properties leading to a wide range of applications in catalysis, electrooptics, magnetism, medicine and biology [46]. POMs are usually synthesized in solution at low temperature. In order to be luminescent, they incorporate, most of the time, emissive ions. The incorporation of guest ions is possible when vacant coordinating sites are present in the POM oxygen-based lattice. Several emissive POM incorporating lanthanide ions have been reported with mostly europium cation as guest [47]. Among them, [EuW10O36]9− anion exhibits the highest luminescent quantum yield [48]. Its molecular structure, presented in Fig. 5, shows the eightfold coordination of Eu3+ by the two [W5O18]6− units [49]. In these luminescent POMs, emission usually occurs from energy transfer between the oxide matrix and the Eu3+ ion in 4f6 configuration. UV irradiation is absorbed by the ligand-to-metal charge transfer transition in the oxide matrix (O → M) and energy is then transferred to the Eu (III) ion which emits its characteristic 5D0 → 7Fj (j = 0–4) transitions [47].
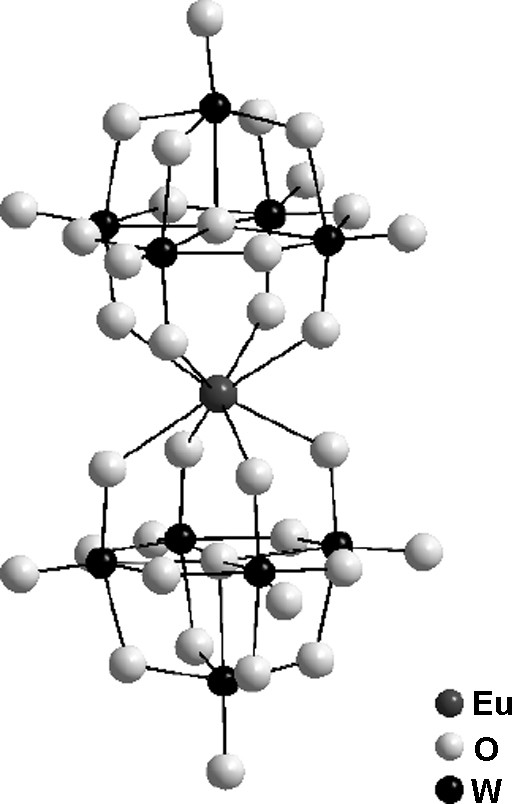
Molecular structure of [EuW10O36]9− polyoxometalate (POM).
Incorporation of [Eu(SiMoxW11−xO39)2]13− (x = 1, 3 and 5) in silica xerogel matrices has been reported to synthesize light emitting materials [50]. The influence of POM composition (parameter x) and of silica matrix nature on the luminescence parameters (emission intensity, lifetime and quantum yield) has been studied for these composite materials. The materials have been synthesized by mixing POM solution with the usual silica precursors (TMOS, PDMS, Glymo) and additives (PEG). Deuterated CH3OD and D2O solvents were used to avoid the POM emission quenching by the OH groups of the matrix that resulted in high values of luminescence lifetime for these materials. Materials incorporating POM with lower x value in silica matrices with higher organic content exhibited the highest emission intensity and quantum yield. On the other hand, stability study in temperature and under UV irradiation showed larger thermal and photochemical degradation compared with the systems with lower organic content. Similar studies have been conducted involving Eu2[TeMo6O24] and [EuW10O36]9− anions and the resulting materials showed enhanced emission intensity and luminescence quantum yield compared with the corresponding POM in solution [51,52].
Transparent and luminescent bulk silica sol–gel materials based on surfactant encapsulated POM has been also synthesized [53]. In order to protect the POM and disperse it in the silica matrix, the usual alkali counter cations of the POM have been this time replaced by functional cationic surfactant. This organic ammonium bears both surfactant alkyl chains and two hydroxyl terminal groups which can copolymerize by condensation with the silica precursors. The resulted surfactant encapsulated [EuW10O36]9− anions were then mixed with TEOS in water and ethanol leading to transparent silica monoliths. Due to the covalent link between the POM and the silica matrix, homogenous distribution of the POM is observed by TEM in contrast with similar materials prepared from Na9[EuW10O36]. Because of the surfactant protection, the structure and fluorescence properties of the POM are well retained in the silica hybrid materials and bright photoluminescence coming from Eu3+ ions can be seen under UV excitation. This study shows that encapsulation using surfactants is an effective method to protect POMs and provide suitable functionalizable sites. This method is also applicable to all kinds of POM in contrast with organic functionalization of the POM anion itself, which usually limits the development of functional materials based on POMs.
Silica nanospheres doped with [Eu(SiMoW10O39)2]13− anions has been synthesized [54]. The POM is incorporated in the silica NPs together with polylysine during the Stöber process. Polylysine has been shown to enhance the POM luminescence in solution by preventing the quenching of the rare-earth emission by water molecules. These luminescent NPs in which the POM is confined at the center of the silica particle show enhanced stability towards water quenching. Application of these emissive NPs as biotags has been considered.
The introduction of the luminescent [(Eu2PW10O38)4(W3O8(H2O)2(OH)4)]22− anion within MCM-41 materials has been reported [55]. MCM-41 is the well-known mesoporous silica-based materials with pores channels assembled in a 2D hexagonal structure with an average diameter of 3 nm. In order to prevent the quenching of the lanthanide luminescence by the silanol groups present at the wall surface of the pores and promoting incorporation of the POM by electrostactic interactions between the matrix, the MCM-41 have been functionalized with amine groups. The incorporation of the POM into the channel of the MCM-41 has been realized by mixing the porous powder in aqueous solution of the POM in weakly acid conditions. XRD and BET measurements respectively show the integrity of the MCM-41 framework after impregnation and the confinement of the POM within the channels. The composite materials exhibit stable and high luminescence with emission bands corresponding to the f–f transitions of the Eu3+ ion.
4.2 Molybdenum halogenide clusters
Octahedral clusters based on d6 and d7 transition metals and formulated [M6X8L6] (M = Mo, W, Re; X = halide or chalcogenide; L = halide or organic coordinating molecule) represent another example of luminescent molecular clusters [56]. These clusters are actually the molecular form of the Chevrel phases M’x[Mo6Q8] (M’ = Pb, Sn, Cu…; X = S, Se, Te) [57] and are prepared by solid state reaction at high temperature. Among this cluster family, the molybdenum chloride member, [Mo6Cl14]2−, presents the highest luminescent quantum yield associated with a long excited state lifetime [58]. The molecular structure of [Mo6Cl14]2− is shown in Fig. 6. The cluster consists in an octahedron of molybdenum (II) atoms with twelve MoMo metallic bonds. Eight chloride atoms bridge each face of the Mo6 octahedron and six more complete the coordination sphere of the molybdenum atoms. Under UV excitation, in solution or in solid state, this cluster emits a red luminescence [59,60] and the origin of this emission has been attributed to transition from a triplet excited state localized on the [Mo6Cl8]4+ cluster core [61].
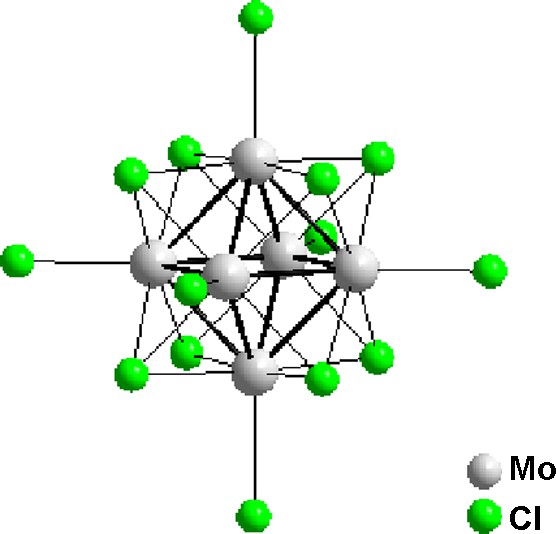
Molecular structure of [Mo6Cl8Cl6]2− cluster.
The first luminescent composite materials based on the incorporation of these clusters in silica-based matrix have been synthesized by hydrolysis of a TMOS solution containing the Mo6Cl12 cluster compound [62]. The resulting materials exhibit the emission and lifetime behavior characteristic of the clusters in solution.
Luminescence of [Mo6Cl14]2− can be efficiently quenched by oxygen. Based on this property, these thermally stable clusters have been studied for the development of real-time, high temperature oxygen sensor based on optical fibers [63]. In this purpose, silica sol–gel films deposited on quartz substrates by dip coating were prepared from TEOS solution containing Mo6Cl12 cluster compound. The luminescence characteristics of films before and after heating at 200 °C were unchanged, indicating that heating did not alter the cluster. Compared to the cluster in solution, quenching by oxygen was smaller in the as-prepared films, but heating at 200 °C increased the quenching, apparently due to increased oxygen permeability resulting from the loss of water or other small molecules from the silica matrix. These results revealed the potential of these clusters as luminophores for oxygen sensors that can operate at relative high temperature compared to organic dyes.
The synthesis of SiO2 NPs incorporating Cs2[Mo6X14] (X = Cl, Br, I) clusters has been reported by the microemulsion method [64]. SEM and TEM analysis indicate an average particle diameter of 45 nm with well-dispersed clusters inside de silica matrix. Slight changes of the clusters luminescence properties have been observed when incorporated inside the silica particles. Indeed, shorter lifetime were observed suggesting interaction between the silica network and the cluster. The broad emission band in the red and near-infrared exhibited by these clusters are potential candidates for biotechnological applications and these composite NPs have been considered for bioimaging and labeling. In a related study, addition of magnetic γ-Fe2O3 nanocrystals to these luminescent [Mo6Br14]2− cluster-silica composites, has lead to bifunctional NPs [65].
4.3 Copper iodide clusters
The d10 coinage metal complexes have been studied for many years due to their rich photoluminescence properties [66–68]. Among them, the tretracopper (I) clusters formulated [Cu4X4L4] (X = Cl, Br, I; L = coordinating molecules) are known to be highly photoluminescent at RT [67,69]. Molecular structure of these cubane type clusters is presented in Fig. 7. These compounds are easily synthesized in solution and can be obtained with different type of ligands (L) allowing their functionalization. Besides, these copper clusters display emission spectra that are strikingly sensitive to their environment, the temperature and the rigidity of the medium. The thermochromic luminescence [70] originates from two emission bands whose relative intensities vary in temperature [71]. At RT, the luminescence is dominated by a low energy band which has been attributed to a combination of a halide-to-metal charge transfer (XMCT) and copper-centered d → s, p transitions. This emission involving a [Cu4I4] cluster centered triplet excited state is essentially independent of the nature of the ligand. At low temperature, this band is extremely weak and the emission is dominated by a higher energy band which has been attributed to a triplet halide-to-ligand charge-transfer (XLCT) excited state [72].

Molecular structure of [Cu4I4(PPh2(CH2)2CH3)4] cluster.
All these properties make these copper clusters particularly attractive to synthesize materials with original optical properties. In this context, our group have reported the synthesis, structural characterizations and optical properties of silica sol–gel films containing [Cu4I4L4] clusters (L = phosphine-based ligands) [73]. The incorporation within the sol–gel silica has been realized with [Cu4I4(PPh2(CH2)2Si(OCH2CH3)3)4] clusters functionalized by alcoxysilane groups able to copolymerize with the silica matrix during the sol–gel process. This allowed a covalent grafting of clusters to the MTEOS silica matrix leading to high concentration and homogeneous distribution of clusters within the matrix. The gel was spin-coated on substrates and transparent and colorless films were obtained. These films exhibit the luminescent properties of the cluster in accordance with XPS and NMR studies demonstrating its integrity in the silica matrix. As shown in Fig. 8, these hybrid silica films show strong yellow emission (λmax = 589 nm) under UV irradiation at RT. By lowering the temperature down to 10 K, a new emission band appeared at higher energy (λmax = 425 nm) which progressively increases in intensity with the concomitant extinction of the yellow band. At 70 K, intensities of the two bands are similar and the addition of blue and yellow light gives the purple emission observed for the film in liquid nitrogen (Fig. 8a). When the sample is progressively warmed up to RT the yellow emission is recovered, indicating a completely reversible thermochromism. Moreover, due to weak Cu–Cu interactions in the cluster, the two emissive states appear as highly coupled with a low energy barrier leading to a clear original isoemissive point and a controlled thermochromism in a large temperature range.
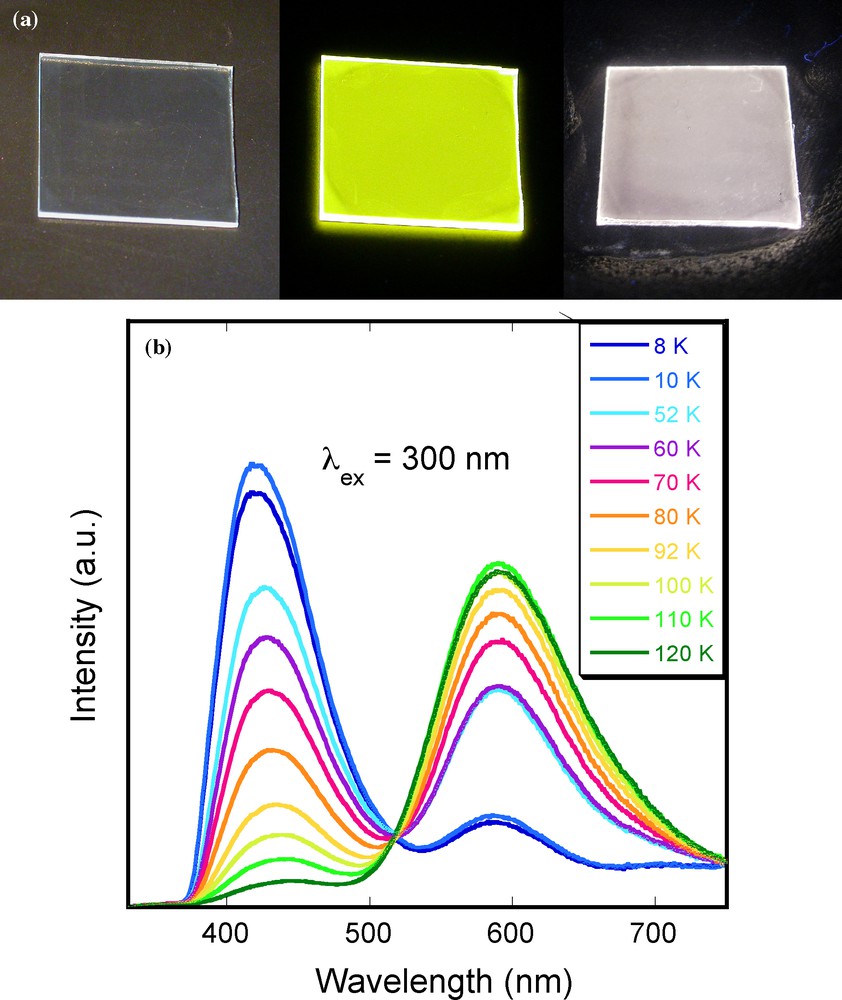
(a): photos of cooper iodide cluster-silica composite films deposited on glass substrate (1.5 × 1.5 cm2). From left to right, under ambient light at room temperature (RT), under UV irradiation at 312 nm (UV lamp) at RT, and under UV irradiation at 312 nm (UV lamp) in liquid nitrogen. (b): temperature dependence of the emission band from 120 to 8 K with λex = 300 nm.
5 Concluding remarks
As shown in this review, the major part of our work concerns the realisation of efficient systems by optimizing intrinsic properties of luminescent inorganic nanophosphors such as the excitation characteristics and the internal luminescence yield. In this field, a crucial point in the future is to shift the excitation wavelength of lanthanide-doped NPs or transition metal clusters towards the blue range to be excited by commercial GaN diodes or towards the infrared range to avoid the photodegradation of biological species. The last property could be obtained by developing new systems able to emit visible light by the up-conversion process, that is by near-infrared light excitation. As example, using the original annealing process of lanthanide-doped-NPs presented here, we have recently succeeded to observe the up-conversion luminescence of Yb,Er-doped YVO4 NPs in water with a strong green emission for an excitation at 980 nm.
For light emission, thin films made from stable luminescent inorganic nanophosphors clearly appear as an alternative to the existing white light emitters such as organic light emitting diodes, polymer light emitting diodes and incandescent light bulbs. In this field of luminescent coatings and beyond the intrinsic properties of luminescent units, a great improvement is expected by varying extrinsic properties of films which can exalt the light emission. A special attention will be put in the future on the different parameters which can influence the dielectric microstructure of the luminescent coatings such as the modulation of the refractive index, the incorporation of diffusive or metallic centres and the realization of multilayer packing.
Acknowledgements
The authors gratefully acknowledge Arnaud Huignard, Valérie Buissette, Domitille Giaume, Geneviève Mialon and Cédric Tard for their crucial contributions to the work discussed here.