

1 Introduction
Phosphines are one of the most important classes of ligands employed in organometallic and coordination chemistry. The ease of fine-tuning the electronic and steric properties of phosphines through variation of the substituents make them ideal ligands for homogeneous catalysts, allowing the reactivity of the catalytic species to be controlled. A wealth of examples of homogeneously catalysed reactions employing metal-phosphine complexes, in particular tertiary phosphine complexes, may be found in the literature [1]. More recent interest in biphasic catalysis has led to the development of water soluble phosphines incorporating polar or ionic functionalities such as amino, hydroxyl, sulphonate or carboxylate groups [2].
Many of the properties that make metal-phosphine complexes attractive for use in catalysis are also relevant for medicinal applications. The reactivity of metal-based drugs must be controlled to prevent undesirable side reactions with amino acids or carbohydrates in the blood stream, while allowing binding to cellular targets through ligand substitution. Furthermore, a balance between hydrophilicity and lipophilicity is necessary for the drug to be soluble in aqueous solution but still able to pass through the phospholipid cell membrane. Finally, electronic properties such as pKa must be carefully tuned; protonation of amino or hydroxyl groups may be essential to the water solubility of a compound but equally, a highly charged compound might not be able to penetrate a cell membrane. On the other hand, protonation in the cell can cause accumulation again by hindering passive diffusion through the cell membrane. While metal phosphine complexes have found applications in many areas of medicinal chemistry, including imaging, anti-inflammatory and antibacterial applications [3], this article will focus on the development of metal-phosphine anticancer agents.
The earliest and most prominent example of a medicinal application of a metal-phosphine compound is the gold(I) complex auranofin [(2,3,4,6-tetra-O-acetyl-1-thio-β-d-glucopyranosato-κS)(triethylphosphine)gold(I)]. In clinical use as an antirheumatic drug, auranofin has also been found to be cytotoxic to leukemia cells [4], prompting the development of a series of gold-triethylphosphine analogues as anticancer agents [5]. Chelate phosphine complexes such as ethylenebis(diphenylphosphine)gold also exhibit excellent antiproliferative activity, though are found to operate through a very different mechanism to the triethylphosphine series [6]. While the efficacy of the initial diphenylethylphosphine complexes was limited by toxicity towards healthy cells, leading to undesirable side effects, replacement of the phenyl groups by 4-pyridyl moieties resulted in very good selectivity towards cancerous cells [7]. By combining a monodentate and a bidentate ligand in the mixed phosphine complex triphenylphosphine[1,3-bis(diphenylphosphino)propane]gold(I) chloride, yielded a highly potent compound (IC50 = 0.01 μM in MCF-7 cell line) [8] (Fig. 1).

Examples of gold-phosphine complexes developed as anticancer agents.
Platinum-based compounds are the most investigated and best studied metal-based anticancer drugs that are in worldwide clinical use. Despite the huge number of platinum compounds that have been developed as anticancer agents, relatively few examples of platinum phosphine complexes have been reported. Romerosa et al. have investigated bisphosphine complexes of formula cis-[Pt(PR3)2Cl2] [9]. The combination of a lipophilic triphenylphosphine ligand and the hydrophilic 1,3,5-triaza-7-phosphatricyclo[3.3.1.1.]decane (pta) ligand proved to be the most cytotoxic, with bis-triphenylphosphine and bis-pta complexes exhibiting negligible antiproliferative activity. Trans phosphine amine complexes investigated by Navarro-Ranninger et al. bearing PPh3 or PPhMe2 ligands were found to induce apoptosis in both cisplatin sensitive and resistant cell lines, representing a promising means of treating cancers where cisplatin is inactive [10].
Of the plethora of new transition metal complexes being developed as potential anticancer agents, ruthenium has emerged as a promising candidate. Attractive features include a rich synthetic chemistry and low general toxicity, attributed to the ability to mimic iron in binding to iron transport proteins. To date, two ruthenium compounds have succeeded to clinical trials, both being Ru(III) coordination complexes with N-donor ligands [11]. However, many different types of compounds have been developed including polynuclear [12], polypyridyl [13] and more recently, organometallic compounds [14].
The anticancer properties of organometallic ruthenium-cyclopentadienyl complexes containing two or three phosphine ligands have been investigated by Romerosa et al. [15,16]. Both hydrophobic and hydrophilic ligands were used, such as pta-Me+, PPh3 or TPPMS (Fig. 2). The degree of interaction with DNA seems to depend on the presence of either a labile leaving group, such as Cl−, which would allow the complex to bind covalently to DNA, or a TPPMS group, which is known to bind DNA through non-covalent interactions.
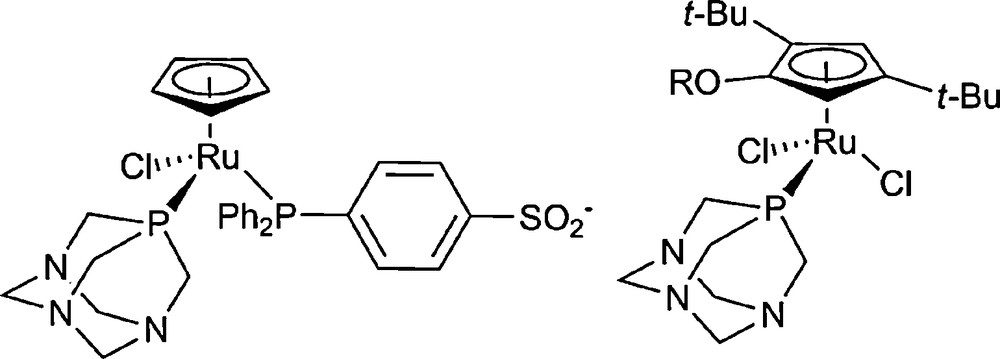
Cyclopentadienyl-ruthenium-phosphine complexes investigated for anticancer activity.
The cytotoxicity of Ru-cyclopentadienyl-pta complexes was found to depend considerably on the nature of the arene ligand. Cp and Cp* complexes of the type [(Cp)RuCl(pta)2] show no activity in the A2780 ovarian cancer cell line and only the Cp complex displays a cytotoxic effect in the TS/A cell line with an IC50 of 100 μM [17]. Remarkably, where a sterically demanding Cp ligand, Cp’OR (η5-1-alkoxy-2,4-di-tert-butyl-3-neopentylcyclopentadienyl, R = Me, Et) is employed, in vitro cytotoxicity is increased by two orders of magnitude (Fig. 2) [18].
Our work has focused on the development of a series of organometallic Ru(II) compounds of the general formula [Ru(η6-arene)Cl2(pta)] (RAPTA), comprising a lipophilic arene ligand, the hydrophilic pta ligand and two labile chlorido ligands (Fig. 3). In vivo the compounds are effective against both primary [19] and metastatic [20] tumours, while exhibiting very low general toxicity. This high selectivity for tumour cells is also evident in vitro, where the series exhibits negligible toxicity in the healthy cell model HBL-100 [20].
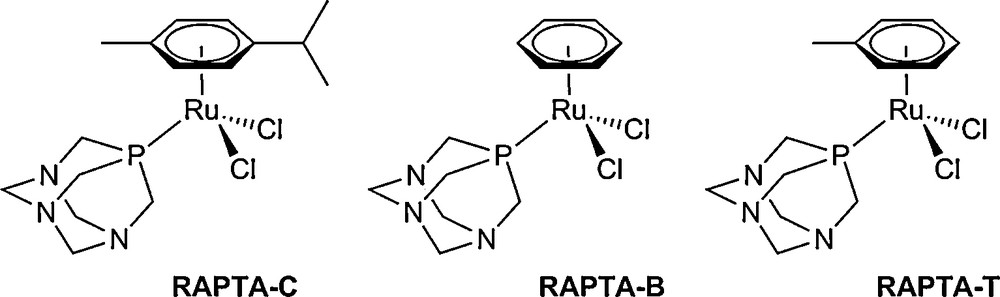
The original RAPTA series, which are active against metastases.
In water, aquation occurs rapidly through loss of a chlorido ligand, generating the labile complex [Ru(η6-arene)Cl(H2O)(pta)]+. In a solution of 100 mM NaCl (equivalent to chloride concentration of the blood stream 100 mM), hydrolysis is suppressed, suggesting that the drug is activated only on reaching the lower salt environment of the cell ([NaCl] = 4 mM). This hypothesis is supported by binding studies with model targets, such as oligonucleotides and proteins, where chlorido ligands are always lost in the formation of Ru-biomolecule adducts [21,22]. Interestingly the rate of hydrolysis has little influence on the reactivity with cellular targets; replacement of the two chlorido ligands with a chelating oxalato ligand renders the complex resistant to hydrolysis, whereas reactivity towards a DNA oligomer is unaffected [23].
Other examples of modulating the ligand sphere have been found to greatly influence properties of the compounds. It was hypothesised that the introduction of polar groups to the arene and pta ligands would improve interactions with DNA through hydrogen bonding and subsequently increase cytotoxicity. A series of compounds with alcohol or amine substituents on the arene ring were investigated, in addition to compounds where the pta ligand had been replaced with dapta (diacetylpta), and their reactivity towards a DNA oligomer and in vitro activity were evaluated. While the polar arene ligands did increase DNA binding, they were significantly less active in vitro, correlated to reduced uptake into the cell. The compounds were also demonstrated to increase cytotoxicity towards the healthy cell model. DAPTA compounds also exhibited lower in vitro activity with respect to their pta analogues, but additionally showed reduced affinity for the DNA oligomer [24].
A similar effect was observed when a chlorido ligand was replaced by a triphenylphosphine group in the monocationic compound [Ru(η6-arene)Cl(PPh3)(pta)]BF4. The hydrophobic group improves uptake into the cell, and subsequently cytotoxicity, and also increases binding to a DNA oligomer. However, the monocationic compounds proved to be much more toxic towards the healthy cell model than their dichlorido analogues, and interestingly also showed reduced binding to model proteins ubiquitin and cytochrome-c [22]. In contrast, replacement of the pta ligand with a chelate analogue, ptn (3,7-dimethyl-7-phospha-1,3,5-triazabicyclo[3.3.1]nonane), yields a complex (RAPTN-C) which is inert in aqueous solution and shows very little affinity for DNA, but binds strongly to the model protein ubiquitin and exhibits superior in vitro cytotoxicity to its pta analogue [25]. The poor correlation between cytotoxicity and DNA binding affinity suggests that DNA may not be the primary target for this class of compounds.
Here we investigate a bis-phosphine complex, [Ru(η6-cymene)(PPh2(o-C6H4O)-κ2-P,O)(pta)]CI, with the aim of combining the improved uptake achieved by incorporation of a triphenylphosphine group with the increased kinetic stability gained through chelate ligands by replacing both chlorido ligands with a strongly chelating phosphine group, ortho-oxy-triphenylphosphine.
2 Results and discussion
The ligand PPh2(o-C6H4OH) is prepared in good yield by Pd-catalysed cross coupling according to a procedure described by Herd et al. [31]. Reaction of two equivalents of phosphine with the ruthenium-cymene chlorido-bridged dimer in the presence of Cs2CO3 yields the half-sandwich, chelate complex, [Ru(η6-cymene)C1(PPh2(o-C6H4O)-κ2-P,O)] [26], which is converted to the chiral monocation [Ru(η6-cymene)(PPh2(o-C6H4O)-κ2-P,O)(pta)]Cl 1, by reaction with a slight excess of pta. Compound 1 is soluble in water and polar organic solvents. The complex was characterised by 1H, 13C and 31P NMR spectroscopy (see Experimental section), and X-ray crystallography. All further studies were conducted using the racemic mixture of 1 (Scheme 1).
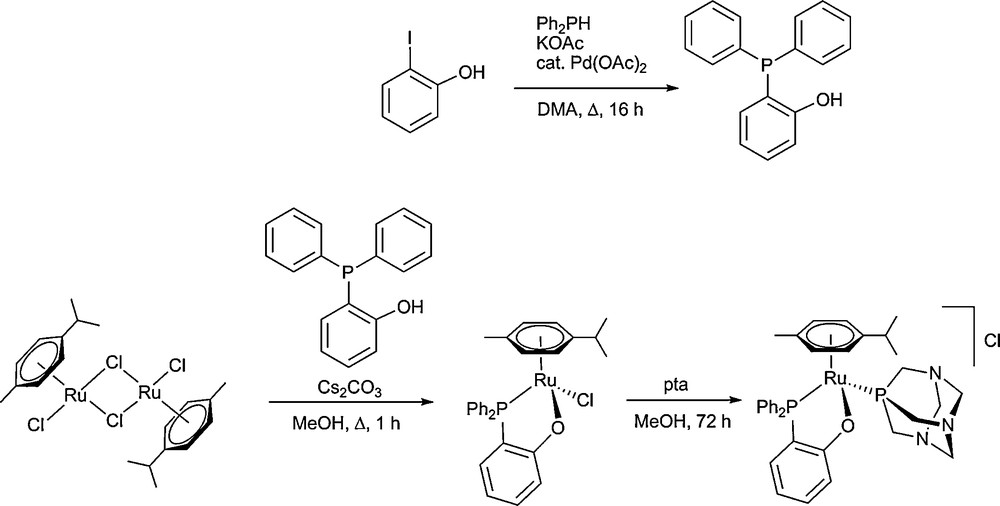
Synthesis of PPh2(o-C6H4OH) and [Ru(η6-cymene)(PPh2(o-C6H4O)-κ2-P,O)(pta)]Cl 1.
2.1 Characterisation in the solid state
Single crystals of 1 suitable for X-ray diffraction were obtained via slow diffusion (see Experimental section for conditions, data collection and refinement details) Table 1. The resulting structure is shown in Fig. 4 and relevant bond parameters are given in the caption. The half-sandwich compound exhibits the typical piano stool structure and is very similar to that of the precursor complex [Ru(η6-cymene)C1(PPh2(o-C6H4O)-κ2-P,O)] [26]. The ruthenium-triarylphosphine bond length Ru1–P1 is shorter than the ruthenium-triarylphosphine bond lengths reported for [Ru(η6-cymene)Cl(PPh3)(pta)]BF4 and [Ru(η6-cymene)Cl(P(C6H4F)3)(pta)]BF4 (2.3177(5) vs. 2.3590(14) and 2.3424(6) Å) [22,27]. In contrast, the Ru–pta bond length is similar to that observed for other RAPTA compounds [28].
Selected crystallographic data for 1.
| Parameter | 1 |
| Formula | C34.50H43Cl2N3O2P2Ru |
| Fw (g mol−1) | 765.63 |
| Crystal system | Triclinic |
| Space group | P-1 |
| a (Å) | 8.8929(3) |
| b (Å) | 10.6189(3) |
| c (Å) | 19.3789(6) |
| α (°) | 105.676(3) |
| β (°) | 91.842(3) |
| γ (°) | 104.19(3) |
| Volume (Å3) | 1698.66(9) |
| Z | 2 |
| Dcalc (g cm−3) | 1.497 |
| μ (mm−1) | 0.697 |
| F(000) | 790 |
| Temp (K) | 140(2) |
| Measured reflections | 15106 |
| Unique reflections | 6625 |
| Theta range (°)/completeness (%) | 3.05 to 26.02/99.4 |
| No. of data/parameters/restraints | 6625/412/0 |
| GooFa | 1.050 |
| Rb [I > 2s(I)] | 0.0246 |
| wR2b (all data) | 0.0643 |
| Largest diff. peak/hole (e.Å−3) | 0.617/−0.633 |
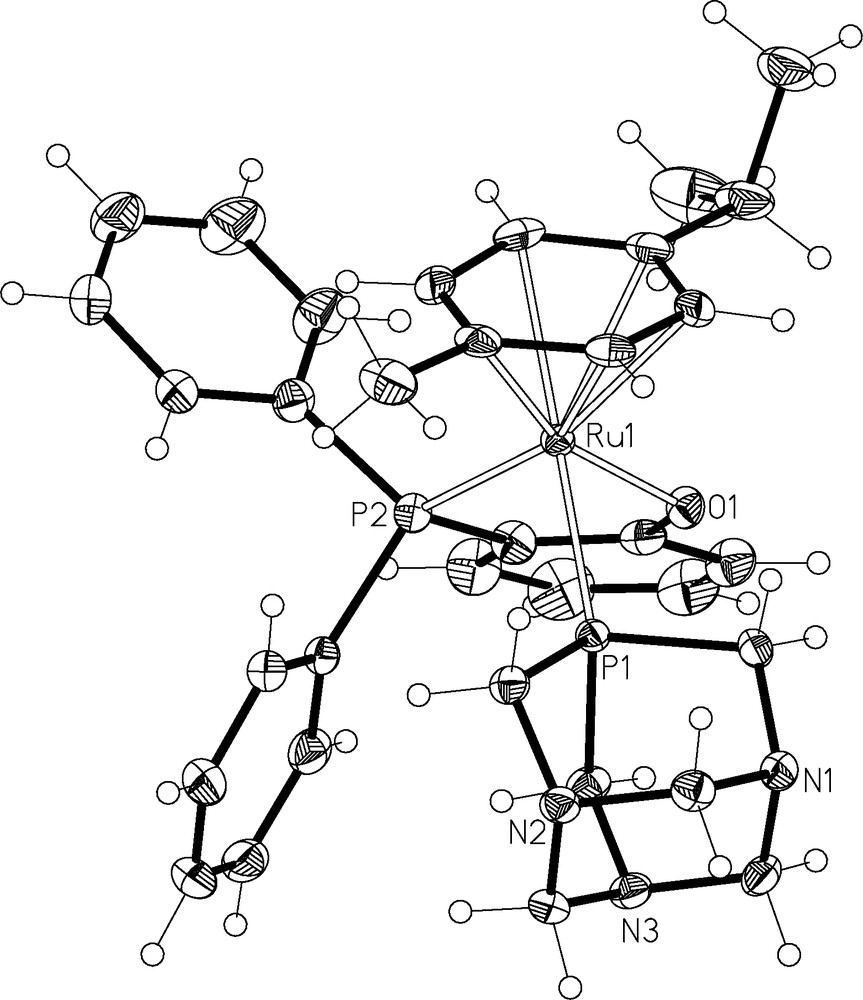
ORTEP representation of 1. Ellipsoids are drawn at 50% probability level and the counter ion and solvate have been omitted for clarity. Key bond lengths (Å) and angles (°) Ru(1)–P(2) 2.2993(5), Ru(1)–P(1) 2.3177(5), Ru(1)–O(1) 2.0933(13), O(1)–Ru(1)–P(1) 82.94(4), O(1)–Ru(1)–P(1) 82.25(4), P(2)–Ru(1)–P(1) 94.848(18). Hydrogen bonds: O(2)–H(2D)…O(1) 2.14(4), O(2)–H(2C)…Cl(1) 2.40(4).
The stability of 1 under physiological conditions was studied using 31P{1H} NMR and UV-VIS spectroscopy. Solutions of 100 μM of metal complex were prepared in water or PBS (pH 7.2) and observed over 7 days. In each case, the spectra remained unchanged. When the pH of a fresh solution of 1 in PBS was gradually lowered by addition of 0.1 M HCl, the 31P{1H} NMR spectrum was unchanged until pH 2, where decomposition was indicated by loss of the original peak and formation of a new species.
It has previously been demonstrated that modifying RAPTA-C by replacement of the chlorido ligands by a bidentate oxalate does not reduce reactivity towards a single stranded DNA oligomer [23]. In contrast, RAPTN-C was significantly less reactive towards a double stranded DNA oligomer than RAPTA-C, though the two compounds showed similar reactivity towards model protein ubiquitin [25]. The reactivity of 1 towards a double-stranded oligomer 5′-GTATTGGCACGTA-3′ was analysed by mass spectrometry and gel electrophoresis. Samples were incubated at 1:1 and 5:1 complex:oligomer ratios and analysed by nESI-FT-ICR-MS after 24 and 72 hours (see Experimental section). In an analogous study, RAPTA-C forms mainly Ru-arene-pta (up to 70% relative intensity) and smaller amounts of Ru-pta adducts (up to 10%, referenced to unreacted single stranded GTATTGGCACGTA, the most abundant peak at charge state −7). For the 5:1 incubation, no assignable signals were present due to oligonucleotide decomposition (confirmed by gel electrophoresis) [25]. However, for 1 no adduct peaks were observed under any conditions with the most abundant peak corresponding to the unreacted oligonucleotide strands.
To supplement the MS study, polyacrylamide gel electrophoresis (PAGE) was performed for all samples (aliquots equivalent to 0.8 μg oligonucleotide as well as standards of 0.8, 0.4 and 0.08 μg of pure oligonucleotide for semiquantitative analysis). In agreement with the MS study, negligible DNA degradation is observed following incubation with 1 under any of the investigated conditions (Fig. 5).
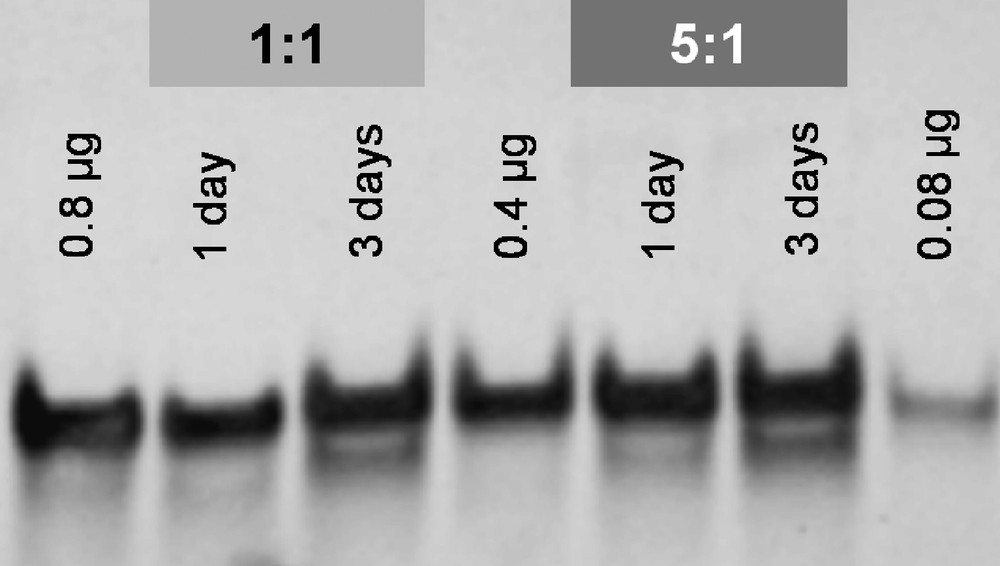
Polyacrylamide gel of 1 incubated with ds(5′-GTATTGGCACGTA-3′) at ratios of 1:1 and 5:1 for 1 and 3 days in water at 37 °C. Standards comprised 0.8, 0.4 and 0.08 μg of unmodified oligonucleotide.
When incubated with ubiquitin under similar conditions (1 or 3 days incubation, 1:1 or 5:1 complex:protein), compound 1 formed only minor adducts with the protein. A species detected at m/z 826 with a relative intensity to the [Ub + 11H]11+ peak of 1.5% was assigned to an [Ub–Ru(η6-cymene)(PPh2(o-C6H4O)–K2–P,O] ion. In contrast, RAPTA-C, RAPTN-C and [Ru(η6-phenyl-ethanol)Cl(PPh3)(pta)]BF4 all bind to ubiquitin efficiently. The absence of a labile leaving group such as a chloride in 1 appears to prevent binding to model cellular targets.
Compound 1 exhibits an IC50 of 138 μM, which was determined using the MTT test on A2780 ovarian cancer cells. Compared to platinum compounds, the compound is only weakly active in vitro, however, other drug candidates with even lower anticancer activity in cell assays exhibit promising in vivo activity and are even in clinical trials (e.g., NAMI-A). In spite of its inertness in aqueous solution and towards cellular targets, 1 proved to be almost 3-fold more active than the parent dichlorido complex RAPTA-C. As 1 appears to be inert under biological and acidic conditions, the possibility that it acts through non-covalent interactions cannot be excluded. Related TPPMS complexes are known to act as DNA intercalators [15,16], and other RAPTA compounds have been found to inhibit enzymes linked to cancer progression, binding in the active site through coordination but also hydrogen bonding and hydrophobic interactions [29].
2.2 Conclusions
The water soluble bis-phosphine complex, 1, was synthesised and its properties compared to other RAPTA complexes. The compound is around 3-fold more cytotoxic than the parent dichlorido complex RAPTA-C, however, unlike other RAPTA compounds, it does not form significant amounts of adducts with either ubiquitin or double-stranded DNA. It is possible that 1 exerts its cytotoxic effect through non-covalent interactions and further studies will be carried out to evaluate its inhibition of relevant enzymes.
3 Experimental
[Ru(η6-p-cymene)Cl2]2, PPh2(o-C6H4OH), [Ru(η6-p-cymene)Cl(PPh2(o-C6H4O)κ2-P,O)] and pta were prepared as previously described [30–32]. All solvents were degassed prior to use and manipulations were carried out using standard Schlenk techniques. 1H and 31P NMR spectra were recorded at 400.13 and 167.1 MHz on a Bruker Avance DPX spectrometer at room temperature. UV-vis spectra were recorded on a JASCO V-550 spectrophotometer.
3.1 Synthesis of [Ru(η6-cymene)(PPh2(o-C6H4O-κ2-P,O)(pta)]Cl (1)
To a methanol solution (100 mL) of (PPh2(o-C6H4O)-κ2-P,O) (500 mg, 0.91 mM), pta (186 mg, 1.2 mM) was added. The solution was stirred at room temperature for 72 h and 31P NMR spectroscopy indicated that the reaction had reached completion. The solvent volume was reduced to ca. 10 mL and the product was precipitated with diethyl ether (10 mL), filtered and washed with diethyl ether (2 × 20 mL) and pentane (2 × 20 mL). Slow diffusion of pentane into a CHCl3 solution gave crystals suitable for X-ray diffraction. Yield: 591 mg (92%) of yellow powder. 1H NMR (CDCl3) δ: 7.59–7.82 (m, 8H, PAr3), 7.38–7.56 (m, 6H, PAr3), 6.10 (d, 3JHH = 5.0 Hz, 2H, Hm), 5.60 (d, 3JHH 5.6 = Hz, 2H, Ho), 4.42 (d, 2JHH = 13.2 Hz, 3H, pta), 4.24 (d, 2JHH = 12.4 Hz, 3H, pta), 3.98 (s, 6H, pta), 2.52–2.67 (m, 1H, CH(CH3)2), 2.22 (s, 3H, arene-CH3), 1.28 (d, 3JHH = 6.7 Hz, 6H, CH(CH3)2). 13C NMR (CDCl3) δ: 133.5 (dd, JPC = 2 Hz, JPC = 1 Hz), 133.2 (d, JPC = 1 Hz), 132.8 (s), 132.1 (d, JPC = 1 Hz), 131.5 (d, JPC = 1 Hz), 131.3 (d, JPC = 21 Hz), 131.2 (m), 119.8 (s), 119.8 (m), 116.6 (s), 72.6 (s), 72.5 (m), 67.0 (s), 51.8 (m), 51.6 (m), 31.5 (s), 22.6 (m), 21.1 (d, JPC = 1 Hz). 31P NMR (CDCl3) δ: 31.46 (2JPP = 53.8 Hz, PAr3), –38.82 (d, 2JPP = 53.9 Hz, pta).
3.2 Protein and (oligo)nucleotide binding studies
3.2.1 Sample preparation
HPLC-purified double-stranded 13-mer oligonucleotide ds(5′-GTATTGGCACGTA-3′) was purchased as an aqueous solution with a concentration of 0.2 mM from A/S Technology (Denmark) and checked by polyacrylamide gel electrophoresis (PAGE) for complete annealing. 1 was dissolved in water (200 μM stock solutions) and immediately incubated with the oligonucleotide at an effective complex:oligonucleotide ratio of 10:10 μM and 50:10 μM in a total volume of 300 μl in Eppendorf vials (500 μl) in a thermomixer (300 rpm; Eppendorf) at 37 °C. Evaporation was minimized by sealing the tubes with parafilm and covering them with several layers of aluminum foil. Aliquots were taken after 24 and 72 h of incubation. All samples were stored at −20 °C until analysis by mass spectrometry or PAGE.
For MS analysis, the aqueous samples were thawed completely and an aliquot of 10 μl was diluted immediately prior to analysis with 40 μl of an 1.25 mM ammonium acetate solution in MeOH/water/n-propanol = 65/10/5 (v/v/v), resulting in the following final spraying conditions: 2 μM oligonucleotide, 1 mM ammonium acetate, MeOH/water/n-propanol = 65/30/5.
For the protein-binding studies, 1 was incubated with ubiquitin (obtained from bovine red blood cells, min. 90%; Sigma) at a molar ratio of 2:1 in water (final protein concentration: 100 μM) and samples were taken after 24 and 72 h incubation at 37 °C. The samples were diluted 1:100 with H2O/CH3CN/HCOOH = 70/30/1 (v/v/v) and immediately analyzed by mass spectrometry.
3.2.2 Mass spectrometry
For electrospray ionization mass spectrometry, the samples were placed into a 96-well plate in an Advion TriVersa™ robot (Advion Biosciences, Ithaca, NY) equipped with a 5.5 μm-nozzle chip. The ESI robot was controlled with ChipSoft v7.2.0 software employing the following parameters:
- • for oligonucleotides:
- ∘ gas pressure 0.40 psi,
- ∘ voltage 1.8–2.0 kV,
- ∘ sample volume 10 μl,
- ∘ negative ion mode;
- • for protein binding studies:
- ∘ gas pressure 0.90 psi,
- ∘ voltage 1.4–1.6 kV,
- ∘ sample volume 25 μl,
- ∘ positive ion mode.
The samples were analyzed using a hybrid ion-trap-FT-ICR mass spectrometer comprising an LTQ XL and an 11 T FT-ICR MS (both ThermoFisher Scientific, Bremen, Germany). The Xcalibur software bundle (Version 2.0.5, ThermoFischer Scientific) was utilized for data acquisition (Tune Plus version 2.2 SP1; ThermoFisher Scientific) and data analysis (Qual Browser version 2.2; ThermoFisher Scientific). Oligonucleotide mass spectra were recorded at a resolution of 75000 at 500 m/z over a range of m/z 350–2000. One scan consisted of 5 microscans, AGC was set to 1 × 106, the maximum injection time of 500 ms was never exceeded, and the spectrum was averaged over at least 50 scans. The mass spectra were recalibrated using the charge distributions of single-stranded TACGTGCCAATAC (charge states 4– to 8–). Protein binding data was collected in ion trap (m/z 400–2000), FT-ICR (m/z 400–2000, resolution of 75000 at m/z 400) and WSIM mode (m/z 770–870; resolution of 40000 at m/z 800). The mass spectra in WSIM mode were recalibrated using the 10+ and 11+ charge states of ubiquitin as internal standards in the positive ion mode.
3.2.3 Gel electrophoresis
Samples were thawed and 10 μl aliquots (0.8 μg oligonucleotide) were mixed with 2 μl of 6X sample buffer and completely loaded on a native 20% polyacrylamide gel. Puc-mix (smallest oligonucleotide: 45 bp) was used as mass ruler in the two outermost lanes and pure double-stranded DNA of sequence ds(GTATTGGCACGTA) was used in amounts of 0.8, 0.4 and 0.08 μg as standards for estimation of the concentration of the oligonucleotide upon incubation with the metal complex. 1X TBE buffer was used as electrolyte and electrophoresis was performed at a constant current (12 mA) for 3–4 h. Gels were stained with ethidium bromide (0.5 μg/ml) for 15–20 min and visualized by UV light irradiation.
3.3 X-ray structure determination
Data for 1 was collected on a KUMA CCD diffractometer system using graphite-monochromated Mo Kα radiation. Data reduction was performed with CrysAlis Red [33]. The structure was solved by direct methods with SHELX-97 [34] and refined by least squares fit on F2 using SHELX-97 [34]. All hydrogen atoms were refined anisotropically with hydrogen atoms placed in geometrically calculated positions and refined using a riding model. Hydrogen atoms that belonged to the water were located on the electron density difference map and the H–O bond lengths and H–O–H angles constrained to reasonable values. Suitable hydrogen acceptors were located using the program Calc-OH [35]. ORTEP3 [36] was used to produce the graphical representation of 1.
3.4 Cell line and culture conditions and cytotoxicity tests (MTT assay)
The human A2780 ovarian cancer cell line was obtained from the European Collection of Cell Cultures (Salisbury, UK). Cells were grown routinely in RMPI medium containing glucose, 5% foetal calf serum (FCS) and antibiotics at 37 °C and 5% CO2.
Cytotoxicity was determined using the MTT assay (MTT = 3-(4,5-dimethyl-2-thiazoyl)-2,5-diphenyl-2H-tetrazolium bromide). Cells were seeded in 96-well plates as monolayers with 100 μL of cell solution (approximately 20,000 cells) per well and preincubated for 24 h in medium supplemented with 10% FCS. The compound was dissolved in the cell medium and serially diluted and 100 μL of drug solution added to each well. The plates were incubated for 72 h then MTT (5 mg/mL solution in phosphate buffered saline) was added to the cells and the plates were incubated for a further 2 h. The culture medium was aspirated and the purple formazan crystals formed by the mitochondrial dehydrogenase activity of vital cells were dissolved in DMSO. The optical density, directly proportional to the number of surviving cells, was quantified at 540 nm using a multiwall plate reader (iEMS Reader MF, Labsystems US) and the fraction of surviving cells was calculated from the absorbance of untreated control cells. Evaluation is based on means from two independent experiments, each comprising three microcultures at each concentration level.
Electronic supporting information available
The crystallographic information file (cif) of complex 1 is available and the coordinates have been deposited at the CCDC, no. 770453.
Acknowledgements
We thank the Swiss National Science Foundation and EPFL for financial support.