

1 Introduction
Isoprene displays a very attractive and inexpensive feedstock for the synthesis of various polymeric materials with distinct chemical, physical, and mechanical properties [1–3]. Homopolymerization of isoprene gives access to polyisoprenes with several microstructures allowing for the production of materials with markedly different characteristics. Nature provides highly stereoregular cis-1,4 polyisoprene as the major component in natural rubber (NR, caoutchouc, > 99% cis content, Mn ≈ 2 × 106 g mol−1) which is the raw material for numerous rubber applications [4]. Further, Gutta-percha produced by Palaquium gutta and several other evergreen trees of East Asia, is an isomer of NR with an all-trans (> 99%) configuration and lower molecular weight (Mn = 1.4–1.7 × 105 g mol−1). While high cis-1,4 polyisoprene acts as an excellent elastomer, high trans-1,4 polyisoprene is characterized as a thermoplastic crystalline polymer with a melting point of 62 °C. The chemical structure of isoprene allows for two more microstructures in the resulting polymers, crystalline iso-3,4 polyisoprene and the less commonly observed 1,2 polymer.
Considering the limited supply of natural rubber and the increasing demand for high-performance synthetic rubbers, the development of catalyst systems producing highly stereoregular polymers and copolymers has grown in importance. The synthesis of highly stereoregular cisPIP with Ziegler-type catalysts is well established [1–3]. In particular, catalyst mixtures with rare-earth metal components such as neodymium represent a prominent class of high-performance catalysts for the industrial stereospecific polymerization of 1,3 dienes [2,3]. Additionally, molecular systems based on lanthanide metallocene and postmetallocene complexes gave access to polymers with very narrow molecular weight distributions and very high cis-1,4 stereoregularity [5–9]. While the cis-specific isoprene polymerization has been studied extensively, the trans-specific polymerization has seen a renewed interest lately [10]. Sterically congested bimetallic rare-earth metal/magnesium allyl and hydroborate species produce polyisoprene with a high trans-1,4 stereoregularity [11–14]. Reports on the 3,4-selective polymerization of isoprene were elusive until the remarkable activity and stereoselectivity of some cationic postmetallocene rare-earth metal alkyl species were reported by Hou and Cui very recently [15].
Controlled crosslinking of polyisoprene or its blendings and copolymerization (e.g., with α-olefins) might be a viable route to afford new high-performance materials. Lately, a number of cationic half-sandwich monoalkyl complexes were found to serve as excellent catalysts for the copolymerization of a range of olefinic monomers such as the syndiospecific copolymerization of styrene with isoprene and the alternating and random copolymerization of isoprene and ethylene [16].
We and Okuda reported on the capability of mono(cyclopentadienyl) bis(tetramethylaluminate) rare-earth metal complexes to initiate the living trans-1,4 stereospecific polymerization of isoprene (trans-1,4 selectivity 99.5%) [17]. To gain fundamental understanding of ancillary ligand, metal size, and cocatalyst effects and hence structure-reactivity relationships in non-metallocene polymerization catalysis, we created a library of half-sandwich bis(tetramethylaluminate) complexes and studied their catalytic performance. Herein, we would like to add tetramethylcyclopentadienyl derivatives [(Cp′)Ln(AlMe4)2]n to this library, emphasizing their synthesis, structural chemistry, cation formation, and catalytic performance in the polymerization of isoprene.
2 Synthesis and structural chemistry of half-sandwich bis(tetramethylaluminate) complexes [(C5Me4H)Ln(AlMe4)2] (2)
Protonolysis reaction of homoleptic complexes [Ln(AlMe4)3] (Ln = Lu (1a), Y (1b), Sm (1c), Nd (1d), and La (1e)) [18] with one equivalent of H(Cp′) (Cp′ = C5Me4H) in hexane at ambient temperature yielded the corresponding bis(tetramethylaluminate) complexes [(Cp′)Ln(AlMe4)2] (2) in good yields (Scheme 1). Instant gas evolution evidenced the anticipated methane elimination reaction and hence, the immediate acid-base reaction of [Ln(AlMe4)3] and the cyclopentadiene (Caution: volatiles containing trimethylaluminum react violently when exposed to air).
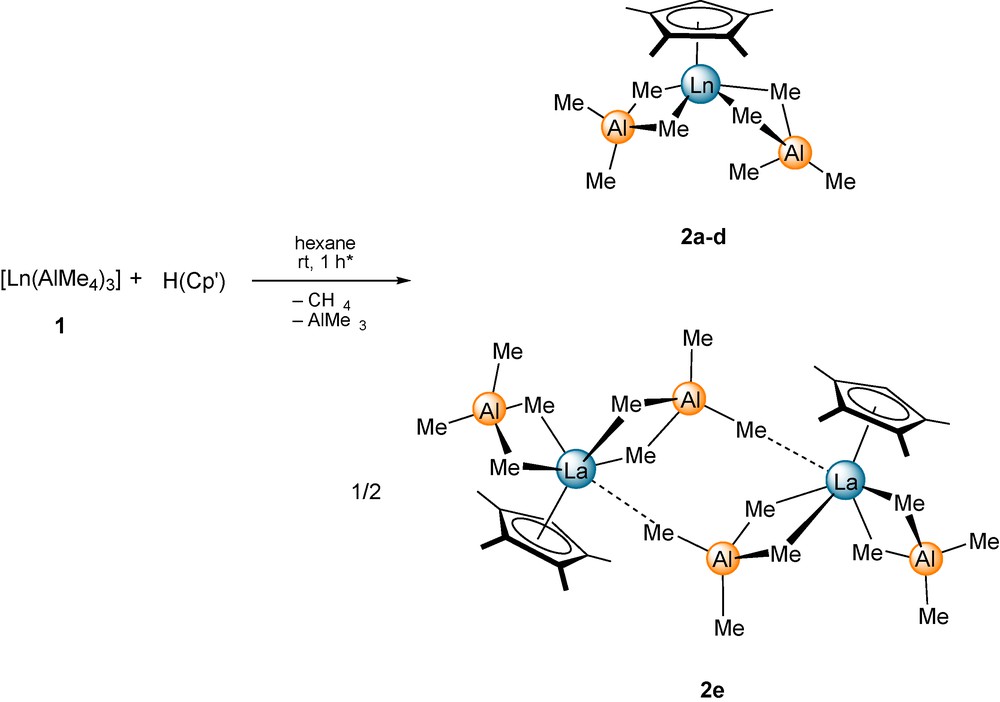
Synthesis of [(Cp′)Ln(AlMe4)2] (2) by methane elimination (Ln = Lu (2a); Ln = Y (2b); Ln = Sm (2c); Ln = Nd (2d); Ln = La (2e)). * For the synthesis of 2a longer reaction times of 16 h are required.
The 1H NMR spectra of the diamagnetic mono(cyclopentadienyl) complexes 2 (Ln = Lu, Y, La) show the expected signal set for the Cp′ ligand and only one narrow signal in the metal alkyl region which can be assigned to the [Al(μ-Me)2Me2] moieties indicating a rapid exchange of bridging and terminal methyl groups. These resonances are slightly shifted to higher field compared to the homoleptic precursors [18]. A signal splitting of the 1H methyl resonance in the yttrium compound 2b (2JYH ≅ 2.0 Hz) is clearly attributable to a two-bond 1H-89Y scalar coupling.
Despite their paramagnetic nature, good quality and informative 1H and 13C NMR spectra could further be obtained for paramagnetic compounds [(Cp′)Sm(AlMe4)2] (2c) and [(Cp′)Nd(AlMe4)2] (2d). The paramagnetic LnIII metal centers influence the 1H and 13C NMR spectra differently, probably due to the varying relaxation behavior of the unpaired electron spins belonging to the paramagnetic metal centers. As for mono(cyclopentadienyl) complexes [(CpR)Ln(AlMe4)2] (Ln = Sm, Nd; CpR = (C5Me5), {1,3-(Me3Si)2C5H3}, (C5Me4SiMe3), {1,2,4-(Me3C)3C5H2}), significant paramagnetic shifts and broadening effects for the 1H and 13C resonances are observed for complexes containing neodymium (1H NMR shift of [AlMe4]: 4.30 ppm), while such effects are less pronounced for the respective samarium compounds (1H NMR shift of [AlMe4]: −3.22 ppm) [17].
Single crystals of [(Cp′)Sm(AlMe4)2] (2c), [(Cp′)Nd(AlMe4)2] (2d), and [{(Cp′)La(AlMe4)2}2] (2e) were grown from saturated hexane solutions at −30 °C. The X-ray crystallographic analyses of the samarium and neodymium derivatives (2c and 2d) revealed structural motifs as found in the solid-state structures of complexes [(CpR)Ln(AlMe4)2] with one [AlMe4] ligand coordinating in the routinely observed planar η2 fashion and the second one showing a bent η2 coordination with an additional short Ln---(μ-Me) contact (Ln1---C4) (Fig. 1) [17,19].
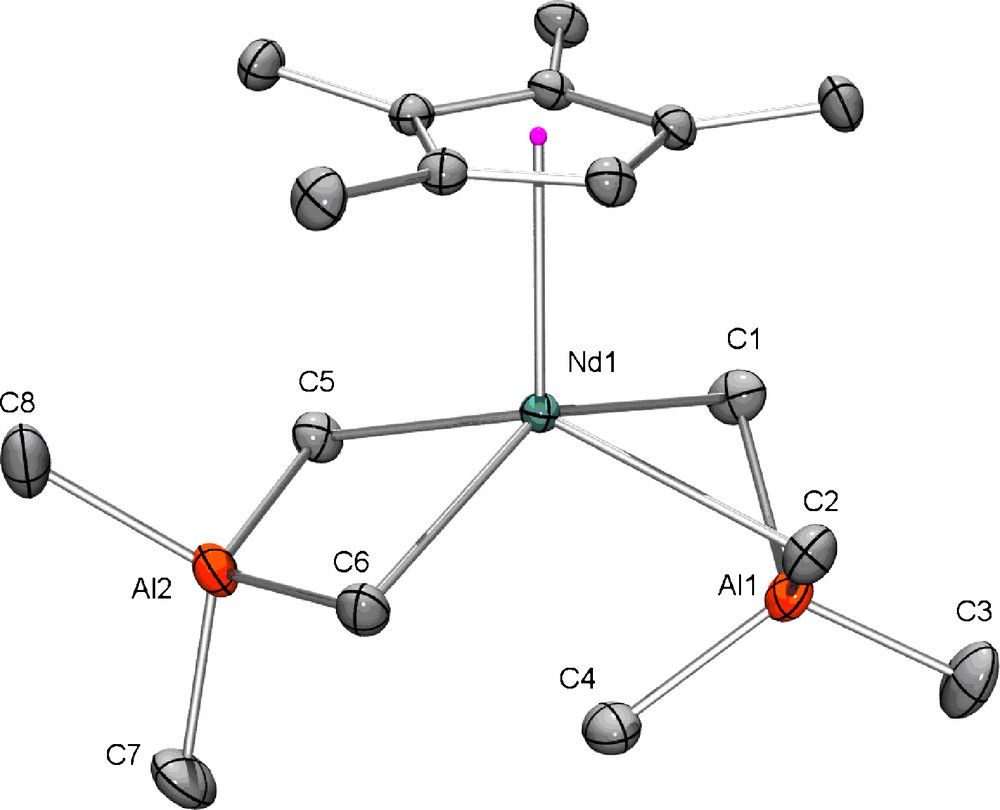
Molecular structure of [(Cp′)Nd(AlMe4)2] (2d), representative of the isostructural complexes 2c and 2d; atomic displacement parameters are set at the 50% level; hydrogen atoms have been omitted for clarity. Selected distances [Å] and angles [°] for 2c: Sm1–C(Cp′) 2.6523(14) - 2.7240(13), Sm1–C1 2.7461(15), Sm1–C2 2.7659(14), Sm1–C5 2.6084(14), Sm1–C6 2.6144(14), Al1–C1 2.0551(16), Al1–C2 2.0586(16), Al1–C3 1.9551(16), Al1–C4 2.0059(17), Al2–C5 2.0869(15), Al2–C6 2.0877(15), Al2–C7 1.9788(18), Al2–C8 1.9763(18), Sm1---Al1 2.9117(4), Sm1---Al2 3.1542(5), Sm1···C4 3.0584(16), C1–Sm1–C2 75.53(5), C5–Sm1–C6 82.09(5), Sm1–C1–Al1 73.07(5), Sm1–C2–Al1 72.58(4), Sm1–C5–Al2 83.62(5), Sm1–C6–Al2 83.45(5), C1–Al1–C2 110.29(7), C1–Al1–C3 110.72(7), C1–Al1–C4 104.33(7), C2–Al1–C3 109.68(7), C2–Al1–C4 104.73(7), C5–Al2–C6 110.47(6), C5–Al2–C7 106.32(7), C5–Al2–C8 110.17(7), C6–Al2–C7 105.76(7), C6–Al2–C8 110.16(7). Selected distances [Å] and angles [°] for 2d: Nd1–C(Cp′) 2.6788(11) - 2.7503(10), Nd1–C1 2.7696(13), Nd1–C2 2.7904(12), Nd1–C5 2.6354(12), Nd1–C6 2.6403(12), Al1–C1 2.0563(13), Al1–C2 2.0588(13), Al1–C3 1.9546(14), Al1–C4 2.0077(15), Al2–C5 2.0832(13), Al2–C6 2.0854(13), Al2–C7 1.9796(15), Al2–C8 1.9749(16), Nd1---Al1 2.9397(4), Nd1---Al2 3.1836(4), Nd1···C4 3.0724(13), C1–Nd1–C2 74.58(4), C5–Nd1–C6 81.13(4), Nd1–C1–Al1 73.39(4), Nd1–C2–Al1 72.88(4), Nd1–C5–Al2 83.99(4), Nd1–C6–Al2 83.82(4), C1–Al1–C2 109.89(6), C1–Al1–C3 110.89(6), C1–Al1–C4 104.48(6), C2–Al1–C3 109.89(6), C2–Al1–C4 105.01(6), C5–Al2–C6 110.77(5), C5–Al2–C7 106.34(6), C5–Al2–C8 109.61(6), C6–Al2–C7 105.77(6), C6–Al2–C8 109.90(6).
As previously shown, such additional Ln---(μ-Me) contacts are significantly affected by the size of the rare-earth metal center and by the steric shielding that is provided by the respective CpR ancillary ligand. Accordingly, the Sm---(μ-Me) contact of 3.0584(16) Å in [(Cp′)Sm(AlMe4)2] (2c) is significantly shorter than the corresponding samarium-methyl contact in [{1,2,4-(Me3C)3C5H2}Sm(AlMe4)2] (3.377(2) Å) featuring a very bulky cyclopentadienyl ligand [17c]. A similar trend was observed for the respective neodymium compounds [(Cp′)Nd(AlMe4)2] (2d) (Ln---(μ-Me): 3.0724(13) Å) and [{1,2,4-(Me3C)3C5H2}Nd(AlMe4)2] (Nd---(μ-Me): 3.326(3) Å) [17c]. The less bulky cyclopentadienyl ligand in [{1,3-(Me3Si)2C5H3}Nd(AlMe4)2], however, enforces a shorter contact of 3.088(2) Å which is comparable to the Nd---(μ-Me) interaction in 2d [17c]. The Ln–C(μ-Me) bond lengths in compounds 2c and 2d increase with increasing LnIII size, the bonds in the bent [AlMe4] ligand being significantly elongated compared to the ones in the planar tetramethylaluminate ligand of the same molecule. The unsubstituted carbon of the cyclopentadienyl ligand (C5Me4H) in both complexes is oriented in between the two [AlMe4] ligands, leaving the methyl substituents orientated above the [AlMe4] ligands.
Contrary to the generally observed monomeric solid-state structures of compounds [(CpR)Ln(AlMe4)2], the X-ray crystallographic investigation of the lanthanum derivative [(Cp′)La(AlMe4)2] (2e) revealed the formation of a so far unprecedented dimeric structure (Fig. 2). The two mono(cyclopentadienyl) units are hereby connected by two [AlMe4] ligands bridging in the rarely observed μ2-η1:η2 fashion. Hexalanthanum cluster [(C5Me5)6La6{(μ-Me)3AlMe}4(μ3-Cl)2(μ2-Cl)6] is the only other crystallographically authenticated compound featuring such a hetero-trinuclear [La(μ-Me2)Al(Me)(μ-Me)La] arrangement [20]. The lanthanum metal centers in complex 2e are further bound by one Cp′ ligand and one additional [AlMe4] coordinating in the commonly observed planar η2 fashion. The La–C(μ-Me) [η2] bonds of the terminal tetramethylaluminate ligands (2.7214(12) Å and 2.7107(11) Å) are comparable to the La–C(μ-Me) bond lengths in [(C5Me5)La{(μ-Me)2AlMe2}2] (2.694(3) Å and 2.802(4) Å) [19b]. In the bridging [AlMe4] ligands, two of the La–C(μ-Me) bonds are significantly elongated. The methyl ligands in trans-position to the electron rich cyclopentadienyl ligand show elongated bonds of 2.9555(11) Å as well as the η1 coordinated methyl group (2.9532(11) Å). Similar separations were found for the La–C(μ-Me) [η1] moieties of cluster compound [(C5Me5)6La6{(μ-Me)3AlMe}4(μ3-Cl)2(μ2-Cl)6] (2.950(3) Å) [20].
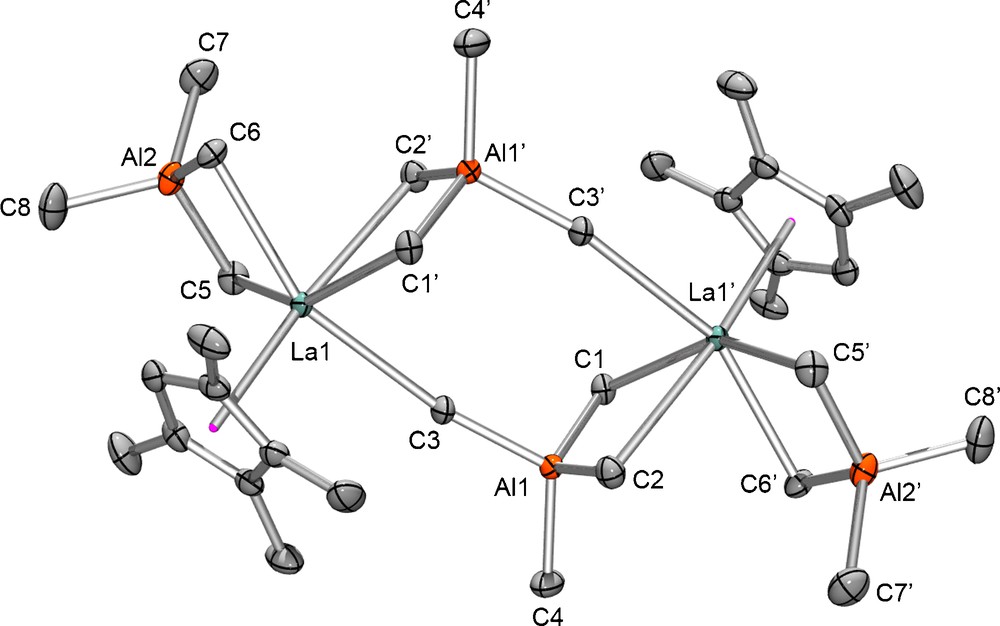
Molecular structure of [{(Cp′)La(AlMe4)2}2] (2e); atomic displacement parameters are set at the 50% level; hydrogen atoms have been omitted for clarity. Selected distances [Å] and angles [°]: La1–C(Cp′) 2.7502(11) - 2.8228(10), La1–C1′ 2.7986(11), La1–C2′ 2.9555(11), La1–C3 2.9532(11), La1–C5 2.7214(12), La1–C6 2.7107(11), Al1–C1 2.0333(11), Al1–C2 2.0382(12), Al1–C3 2.0226(11), Al1–C4 1.9695(12), Al2–C5 2.0823(13), Al2–C6 2.0705(12), Al2–C7 1.9831(14), Al2–C8 1.9750(14), La1---Al1′ 3.4181(4), La1---Al2 3.2570(4), La1---Al1 4.9356(5). C1′–La1–C2′ 72.83(3), C1′–La1–C3 84.57(3), C2′–La1–C3 76.58(3), C5–La1–C6 78.47(3), La1′–C1–Al1 88.58(4), La1′–C2–Al1 84.26(4), La1–C3–Al1 165.31(6), La1–C5–Al2 84.25(4), La1–C6–Al2 84.75(4), C1–Al1–C2 114.19(5), C1–Al1–C3 103.47(5), C1–Al1–C4 109.88(6), C2–Al1–C3 105.99(5), C2–Al1–C4 107.70(6), C3–Al1–C4 115.76(5), C5–Al2–C7 105.90(6), C5–Al2–C8 110.53(6), C6–Al2–C7 105.67(6), C6–Al2–C8 106.42(6), Al1′---La1---Al2 105.151(10), La1---Al1---La1′ 107.94(1). Symmetry operator [−x, −y + 1, −z].
3 Reactivity of [(Cp′)Ln(AlMe4)2] toward [Ph3C][B(C6F5)4], [PhNMe2H][B(C6F5)4], and B(C6F5)3
We previously reported on the reactivity of mono(cyclopentadienyl) bis(tetramethylaluminate) complexes [(CpR)Ln(AlMe4)2] toward perfluorinated organoboron reagents and the superb performance of such catalyst mixtures in the stereospecific polymerization of isoprene [17b,c]. Accordingly, we conducted small-scale equimolar reactions of complexes [(Cp′)Ln(AlMe4)2] (2) with [Ph3C][B(C6F5)4] (A), [PhNMe2H][B(C6F5)4] (B), and B(C6F5)3 (C) as solutions in [D6]benzene in Teflon-valved NMR tubes. The NMR signals for 2 disappeared instantly and the quantitative formation of Ph3CMe and one equivalent of AlMe3 or the quantitative formation of PhNMe2, one equivalent of AlMe3, and CH4, respectively, were observed (Scheme 2). New signals for the Cp′ ligand appeared shifted to slightly higher field in accordance with a stronger coordination toward the highly electron-deficient rare-earth metal cation. High-field shifts were also observed for the signals of the remaining [AlMe4] ligand. The stability of the cationic species 3, however, significantly depends on the size of the rare-earth metal cation (La ≫ Nd > Y > Lu). Ion pairs [(Cp′)La(AlMe4)][B(C6F5)4] (3e), obtained from 2e and A or B are stable for several hours enabling more detailed NMR spectroscopic investigations.
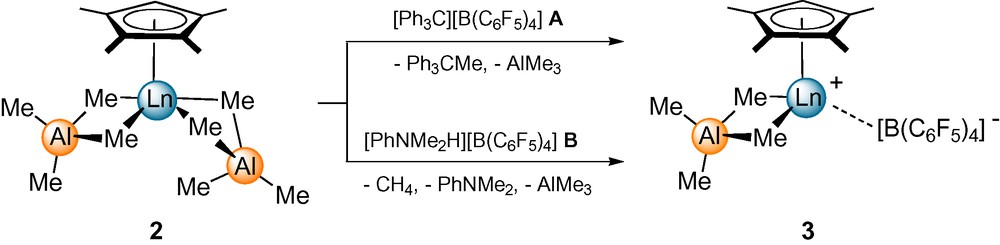
Proposed reaction of [(Cp′)Ln(AlMe4)2] with [Ph3C][B(C6F5)4] (A) and [PhNMe2H][B(C6F5)4] (B) (Ln = Lu (3a), Ln = Y (3b), Ln = Nd (3d), Ln = La (3e). (Note: for 2e the monomeric structure is depicted for simplification).
The 11B NMR spectra of 2e/A and 2e/B in [D6]benzene revealed a broad resonance at δ = −16.2 ppm and −16.3 ppm, respectively. Additionally, the 19F chemical shift differences for the p- and m-F atoms of 4.7 ppm (2e/A) and 4.4 ppm (2e/B) suggest the existence of tight ion pairs [21]. Broad singlets at −0.36 ppm (2e/A) and −0.50 ppm (2e/B) in the 1H NMR spectra integrating to 12 protons clearly correspond to the presence of only one [AlMe4] ligand. Moreover, the chemical shifts of PhNMe2 formed upon reaction of 2e with [PhNMe2H][B(C6F5)4] (B) are not consistent with the reported 1H NMR shifts for free PhNMe2 in [D6]benzene [22]. Particularly, the methyl resonance at 2.24 ppm (2e/B) is shifted to higher field compared with free PhNMe2 (2.50 ppm) suggesting a coordination of N,N-dimethylaniline to the newly formed ion-pair.
Monitoring the reaction of [{(Cp′)La(AlMe4)2}2] (2e) with two equivalents of Lewis acidic B(C6F5)3 (C) in [D6]benzene by 1H NMR spectroscopy revealed the quantitative formation of a new species with signals for the Cp′ ligand shifted to higher field. The 1H NMR spectrum closely resembles the spectrum of the contact ion-pair [{[(Cp*)La{(μ-Me)2AlMe(C6F5)}][Me2Al(C6F5)2]}2] (Cp* = C5Me5) formed in the reaction of [(Cp*)La(AlMe4)2] (5e) and B(C6F5)3 [17b]. The presence of one equivalent of BMe3, the appearance of two aluminum containing species in the 27Al NMR spectrum, and the shock sensitive nature of mixtures 2e/C at higher concentrations further suggest the formation of a cationic species similar to the dimeric contact ion-pair obtained by [(Cp*)La(AlMe4)2] and C (Scheme 3). Two sets of C6F5 resonances appear in the 19F NMR spectrum at 25 °C which can be assigned to a {Me2Al(C6F5)2} and a {AlMe3(C6F5)} unit, respectively. Contrary to the fluxional solution structure observed for mixtures [(Cp*)La(AlMe4)2]/C, an additional La–ortho-fluorine interaction, evidenced by an upfield 19F signal (−124.1 ppm), is present in the cationic species formed by [{(Cp′)La(AlMe4)2}2] and C.
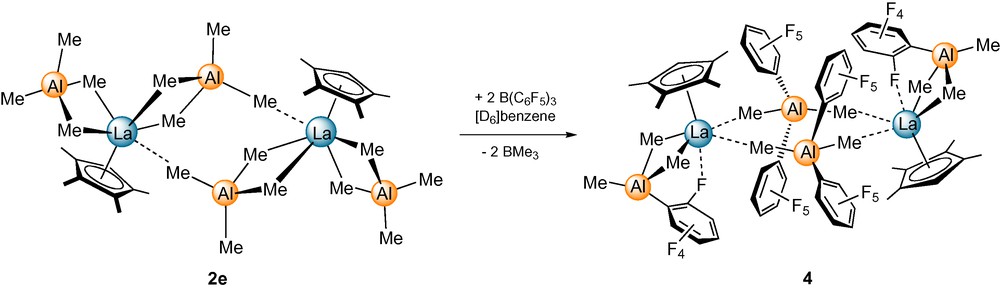
̈Proposed reaction of [{(Cp′)La(AlMe4)2}2] (2e) with B(C6F5)3 (C).
While the reaction of [(Cp*)La(AlMe4)2] and B(C6F5)3 occurred instantly, the analogous reaction of dimeric [{(Cp′)La(AlMe4)2}2] and the Lewis acid is only completed after 3 h. This observation points toward the initial cleavage of the dimeric structure as the rate-limiting step.
4 Polymerization of isoprene
Our previous studies on the catalytic performance of mono(cyclopentadienyl) bis(tetramethylaluminate) complexes [(CpR)Ln(AlMe4)2] demonstrated their potential as initiators for the controlled polymerization of isoprene [17]. Mixtures of the half-sandwich complexes and fluorinated borate or borane reagents provided catalysts with remarkable activity and very high trans-1,4 selectivity (trans-1,4 content up to 99.5%). To extend the series and gain fundamental information on the ancillary ligand influences on the catalyst performance, the herein introduced tetramethylcyclopentadienyl complexes [(Cp′)Ln(AlMe4)2] (2) were employed as precatalysts in the homopolymerization of isoprene. The polymerization results are summarized in Table 1.
Effect of Ln size, cocatalyst, and temperature on the polymerization of isoprene.
| Entrya | Precatalyst | Cocatalystb | t [h] | T [°C] | Yield [%] | Microstructured | Mne (×105) | Eff.f | |||
| trans-1,4- | cis-1,4- | 3,4- | |||||||||
| 1 | [{(Cp′)La(AlMe4)2}2] (2e) | A | 2 | rt | > 99 | 90.5 | 3.5 | 6.0 | 0.7 | 1.10 | 1.00 |
| 2 | [{(Cp′)La(AlMe4)2}2] (2e) | B | 2 | rt | > 99 | 80.8 | 9.8 | 9.3 | 0.6 | 1.07 | 1.06 |
| 3 | [{(Cp′)La(AlMe4)2}2] (2e) | C | 24 | rt | 36 | 93.4 | 4.8 | 1.8 | 2.0 | 1.26 | 0.12 |
| 4 | [{(Cp′)La(AlMe4)2}2] (2e) | C | 24 | 50 | 18 | 91.4 | 6.1 | 2.5 | 1.2 | 1.18 | 0.10 |
| 5 | [{(Cp′)La(AlMe4)2}2] (2e) | C | 48 | rt | 29 | 75.2 | 20.3 | 4.5 | 1.3 | 1.40 | 0.15 |
| 6 | [(Cp′)Nd(AlMe4)2] (2d) | A | 2 | rt | > 99 | 76.3 | 13.0 | 10.7 | 0.5 | 1.12 | 1.36 |
| 7 | [(Cp′)Nd(AlMe4)2] (2d) | B | 2 | rt | > 99 | 79.2 | 13.2 | 7.6 | 0.5 | 1.10 | 1.47 |
| 8 | [(Cp′)Nd(AlMe4)2] (2d) | C | 2 | rt | 76 | 13.5 | 75.1 | 11.4 | 2.9 | 1.52 | 0.18 |
| 9 | [{(Cp′)La(AlMe4)2}2] (2e) | C | 16 | −30c | 0 | – | – | – | – | – | – |
| 10 | [(Cp*)La(AlMe4)2] (5e) | C | 16 | −30c | 0 | – | – | – | – | – | – |
| 11 | [(Cp*)Nd(AlMe4)2] (5d) | C | 16 | −30c | 0 | – | – | – | – | – | – |
| 12 | [(Cp*)La(AlMe4)2] (5e) | A | 16 | −30c | 95 | 95.2 | 1.0 | 3.8 | 1.0 | 1.15 | 0.63 |
| 13 | [(Cp*)La(AlMe4)2] (5e) | B | 16 | −30c | > 99 | 91.1 | 2.7 | 6.4 | 0.9 | 1.08 | 0.75 |
| 14 | [(Cp*)Nd(AlMe4)2] (5d) | A | 16 | −30c | > 99 | 89.3 | 1.8 | 8.9 | 0.9 | 1.20 | 0.76 |
| 15 | [(Cp*)Nd(AlMe4)2] (5d) | B | 16 | −30c | 90 | 64.1 | 13.5 | 22.4 | 0.4 | 1.04 | 1.46 |
| 16 | [(Cp′)Nd(AlMe4)2] (2d) | A | 16 | −30c | > 99 | 77.5 | 13.2 | 9.3 | 0.4 | 1.07 | 1.57 |
| 17 | [(Cp′)Nd(AlMe4)2] (2d) | B | 16 | −30c | > 99 | 54.6 | 29.0 | 16.4 | 0.5 | 1.09 | 1.27 |
| 18 | [{(Cp′)La(AlMe4)2}2] (2e) | A | 16 | −30c | > 99 | 95.2 | 1.9 | 2.9 | 0.8 | 1.05 | 0.90 |
| 19 | [{(Cp′)La(AlMe4)2}2] (2e) | B | 16 | −30c | > 99 | 87.7 | 7.0 | 5.3 | 0.8 | 1.05 | 0.82 |
a Conditions: 0.02 mmol precatalysts, [Ln]/[cocat] = 1:1, 8 mL toluene, 20 mmol isoprene.
b Cocatalyst: A = [Ph3C][B(C6F5)4], B = [PhNMe2H][B(C6F5)4], C = B(C6F5)3; catalyst preformation 20 min at 40 °C.
c Catalyst preformation 20 min at −30 °C.
d Determined by 1H and 13C NMR spectroscopy in CDCl3.
e Determined by means of size-exclusion chromatography (SEC) with a triple detection array.
f Initiation efficiency = Mn(calculated)/Mn(measured).
Cationic species generated in situ upon treatment of [(Cp′)Nd(AlMe4)2] (2d) and [{(Cp′)La(AlMe4)2}2] (2e) with [Ph3C][B(C6F5)4] (A) or [PhNMe2H][B(C6F5)4] (B), respectively, showed excellent activities in the polymerization reactions (Table 1, runs 1, 2, 6, and 7). The observed catalyst activities are comparable to the ones reported for mixtures of [(CpR)Ln(AlMe4)2] and A/B [17b,c]. The high trans-1,4 contents of the resulting polyisoprenes are also in line with the literature reports. They, however, do not exceed a maximum of 90.5% for 2e/A (Table 1, run 1). All polymers show a very narrow molecular weight distribution and for the lanthanum-based catalyst the efficiencies are very close to 100%.
In accordance with a different activation mechanism, the use of B(C6F5)3 (C) as an activator for complexes 2e and 2d resulted in the formation of a catalytically active species with a markedly different performance. Active species formed in mixtures 2e/C and 2d/C polymerize isoprene with low activities compared with the borate activated systems (Table 1, runs 3 and 8). Very high trans-1,4 selectivity was observed for the largest metal center lanthanum (trans-1,4 content 93.4% - for comparison trans-1,4 content for mixtures [(CpR)La(AlMe4)2]/C: CpR = {1,3-(Me3Si)2C5H3}, 89.3%; CpR = {1,2,4-(Me3C)3C5H2}, 90.0%; CpR = (C5Me4SiMe3), 95.6%; CpR = (C5Me5), 99.5%) [17b,c]. While all previously reported catalyst mixtures [(CpR)La(AlMe4)2]/C produced polyisoprene in quantitative yield, the combination of [{(Cp′)La(AlMe4)2}2] (2e) and B(C6F5) (C) reproducibly gave a polymer yield of < 40% (Table 1, run 3). Heating the reaction mixture and prolonged reaction times did not increase the yield of polyisoprene (Table 1, runs 4 and 5). At present the cause of this low polymer yield remains unclear, but the low rate of the cation formation (vide supra) and a rate limiting first insertion of a monomer into a stable La-methyl bond of 4 might be held responsible. Higher polymer yield was obtained with the neodymium derivative [(Cp′)Nd(AlMe4)2] (2d) activated with C (76%, Table 1, run 8). Surprisingly, the representative of the smaller metal center produced polyisoprene with 75.1% cis-1,4 content. Formation of homoleptic [Nd(AlMe4)3] due to ligand redistribution reactions might explain the dramatic selectivity change [8b].
Comparing the catalytic results of our cationic mono(tetramethylaluminate) half-sandwich complexes 2 with the analogous systems featuring a (trimethylsilyl)methyl actor ligand ([(Cp′)Sc(CH2SiMe3)][B(C6F5)4]) reported by Hou, demonstrates the striking trans-1,4 directing effect of the tetramethylaluminate ligand and the influence of the rare-earth metal size. Under comparable reaction conditions such systems produced polyisoprenes with cis-1,4/3,4 microstructures [16e].
Further, the effect of the polymerization temperature on the catalytic performance of [(Cp′)Nd(AlMe4)2] (2d), [{(Cp′)La(AlMe4)2}2] (2e), [(Cp*)Nd(AlMe4)2] (5d), and [(Cp*)La(AlMe4)2] (5e) were investigated. At −30 °C, mixtures of the named precatalysts and B(C6F5)3 (C) did not provide active catalysts (Table 1, runs 9–11). Reasonably, the rates for the catalyst preformation as well as the rates of the first monomer insertion are dramatically reduced at low temperature preventing the formation of polymeric material. In contrast, the same precatalysts activated with the perfluorinated borates [Ph3C][B(C6F5)4] (A) and [PhNMe2H][B(C6F5)4] (B) at −30 °C formed catalytically active species (Table 1, runs 12–19). Generally, all mixtures very efficiently produced polyisoprene with narrower molecular weight distributions than at ambient temperature. As anticipated, catalyst activities were significantly reduced but all catalysts converted the monomer completely or nearly completely into polyisoprene within 16 h. Cooling of the catalyst mixtures significantly increased the trans-1,4 content of the resulting polymers. Particularly, the half-sandwich complexes based on the largest metal center lanthanum (2e and 5e) activated with [Ph3C][B(C6F5)4] (A) performed with a trans-1,4 selectivity of 95.2%1 (Table 1, runs 12 and 18).
The catalytic performance of the catalyst systems based on neodymium complexes 2d and 5d revealed a strong dependence on the applied cocatalyst A or B when cooled to −30 °C (Table 1, runs 14–17). While [Ph3C][B(C6F5)4] (A) as activator resulted in the production of polyisoprene with significantly increased trans-1,4 content (for 5d/A: 69.7% at rt vs. 89.3% at −30 °C), the use of [PhNMe2H][B(C6F5)4] (B) led to a remarkably reduced trans-1,4 selectivity (for 2d/A: 79.2% at rt vs. 54.6% at −30 °C). Possibly, a stronger coordination of PhNMe2 (produced upon cation formation) to the neodymium metal center at low temperatures can be held responsible for the observed selectivity loss.
5 Conclusions
Donor-solvent free half-sandwich complexes [(Cp′)Ln(AlMe4)2]n were synthesized as a follow-up to the library of mono(cyclopentadienyl) bis(tetramethylaluminate) complexes, which has been developed by us recently. X-ray structure analyses of complexes [(Cp′)Ln(AlMe4)2]n (Ln = Sm, Nd, La) revealed an unprecedented dimeric structure of the respective lanthanum compound in the solid state. The slight modification of the ancillary ligands’ sterical demand (C5Me4H vs. C5Me5) not only affects the structural chemistry of the resulting half-sandwich complex but has also significant implications for the catalytic performance. While the contact ion pair formed by [(C5Me5)La(AlMe4)2] and B(C6F5)3 quantitatively produced polyisoprene with an extremely high trans-1,4 content of 99.5%, reduction of the steric bulk at the cyclopentadienyl ancillary ligand by only one methyl group led to a loss of stereocontrol and catalytic activity in mixtures [{(C5Me4H)La(AlMe4)2}2]/B(C6F5)3 (trans-1,4 content 93.4%).
6 Experimental section
6.1 General considerations
All operations were performed under inert atmosphere, using standard Schlenk, high-vacuum, and glovebox techniques (MBraun MBLab; < 1 ppm O2, < 1 ppm H2O). Hexane and toluene were purified using Grubbs columns (MBraun SPS, solvent purification system) and stored in a glovebox. [D6]Benzene was obtained from Aldrich, degassed, dried over Na for 24 h, and filtered. H(C5Me4H), H(C5Me5), and AlMe3 were purchased from Aldrich and used as received. [Ph3C][B(C6F5)4], [PhNMe2H][B(C6F5)4], and B(C6F5)3 were purchased from Boulder Scientific Company and used without further purification. Homoleptic [Ln(AlMe4)3] [18] (Ln = Lu, Y, Sm, Nd, La) and [(C5Me5)Ln(AlMe4)2] [19] (Ln = Nd, La) were prepared according to literature methods. Isoprene was obtained from Aldrich, dried several times over activated molecular sieves (3 Ǻ) and distilled prior to use. The NMR spectra of air sensitive compounds were recorded using Teflon-valved NMR tubes at 25 °C on a Bruker-BIOSPIN-AV500 (5 mm BBO, 1H: 500.13 MHz; 13C: 125.77 MHz) and a Bruker-AVANCE-DMX400 (5 mm BB, 1H: 400.13 MHz; 13C: 100.62 MHz). 1H and 13C shifts are referenced to internal solvent resonances and reported in parts per million relative to TMS. Elemental analyses were performed on an Elementar Vario EL III. The molar masses ( and Mn) of the polymers were determined by size-exclusion chromatography (SEC) in thf at 30 °C. Sample solutions (1.0 mg polymer per mL thf) were filtered through a 0.2 μm syringe filter prior to injection. The chromatographic system (GPCmax, Viscotek) consisted of a GPCmax apparatus (comprising isocratic pump, autosampler, and degassing unit) and a model TDA 302 triple detector array detector comprising refractive index (RI), differential pressure (DP), low angle (7°) light scattering (LALS), and right angle (90°) light scattering (RALS) detection with an integrated column oven and a model 2501 UV detector. Both the dn/dc and the dA/dc data were determined by means of the integrated OmniSec™ software. The flow rate was 1.0 mL min−1. The microstructure of the polyisoprenes was examined via 1H and 13C NMR experiments on the AV500 and the DMX400 spectrometer in [D]chloroform at ambient temperature.
6.2 General procedure for the preparation of [(C5Me4H)Ln(AlMe4)2] (2)
In a glovebox [Ln(AlMe4)3] (1) was dissolved in hexane (2 mL) and H(C5Me4H) (1 equiv) was added dropwise to the alkylaluminate solution under vigorous stirring. Upon the addition, instant gas formation was observed. After the reaction mixture had been stirred for 1 h at ambient temperature, the solvent was removed in vacuo to give 2 as crystalline solids. Crystallization from a solution in hexane at −35 °C gave high yields of single crystals of 2 suitable for X-ray diffraction analysis.
6.3 [(C5Me4H)Lu(AlMe4)2] (2a)
Following the procedure described above but stirring the reaction mixture for 16 h, [Lu(AlMe4)3] (1a) (165 mg, 0.38 mmol) and H(C5Me4H) (46 mg, 0.38 mmol) yielded 2a (104 mg, 0.22 mmol, 58%) as colorless crystals. 1H NMR (400 MHz, [D6]benzene): δ = 5.51 (s, 1H, CH), 1.86 (s, 6H, Me), 1.66 (s, 6H, Me), −0.18 (s, 24H, AlMe4) ppm. 13C NMR (100 MHz, [D6]benzene): δ = 122.1, 121.8, 113.0, 13.8, 11.4, 1.2 (s br, AlMe4) ppm. Elemental Analysis (%) calcd. for C17H37Al2Lu (470.41): C 43.41, H 7.93; found: C 43.30, H 7.94.
6.4 [(C5Me4H)Y(AlMe4)2] (2b)
Following the procedure described above, [Y(AlMe4)3] (1b) (139 mg, 0.40 mmol) and H(C5Me4H) (88 mg, 0.72 mmol) yielded 2b (104 mg, 0.27 mmol, 68%) as colorless crystals. 1H NMR (500 MHz, [D6]benzene): δ = 5.58 (s, 1H, CH), 1.82 (s, 6H, Me), 1.65 (s, 6H, Me), −0.33 (d, JYH = 2.03 Hz, 24H, AlMe4) ppm. 13C NMR (126 MHz, [D6]benzene): δ = 123.5, 123.2, 114.2, 13.8, 11.4, 0.4 (d, JYC = 35.6 Hz, AlMe4) ppm. Elemental Analysis (%) calcd. for C17H37Al2Y (384.34): C 53.12, H 9.70; found: C 53.61, H 10.17.
6.5 [(C5Me4H)Sm(AlMe4)2] (2c)
Following the procedure described above, [Sm(AlMe4)3] (1c) (210 mg, 0.51 mmol) and H(C5Me4H) (68 mg, 0.56 mmol) yielded 2c (107 mg, 0.24 mmol, 47%) as red crystals. 1H NMR (500 MHz, [D6]benzene): δ = 11.12 (s, 1H, CH), 0.82 (s, 6H, Me), 0.37 (s, 6H, Me), −3.22 (s, 24H, AlMe4) ppm. 13C NMR (126 MHz, [D6]benzene): δ = 118.9, 118.7, 111.0, 17.7, 16.1, −20.6 (br. s, AlMe4) ppm. Elemental Analysis (%) calcd. for C17H37Al2Sm (445.80): C 45.8, H 8.4; found: C 45.8, H 8.0.
6.6 [(C5Me4H)Nd(AlMe4)2] (2d)
Following the procedure described above, [Nd(AlMe4)3] (1d) (224 mg, 0.55 mmol) and H(C5Me4H) (72 mg, 0.59 mmol) yielded 2d (242 mg, 0.55 mmol, > 99%) as blue crystals. 1H NMR (500 MHz, [D6]benzene): δ = 11.05 (s, 6H, Me), 9.89 (s, 6H, Me), 4.96 (s, 1H, CH), 4.30 (s, 24H, AlMe4) ppm. 13C NMR (126 MHz, [D6]benzene): δ = 247.2, 238.1, 31.8, 30.1, 23.1, 14.2, −19.3, −21.1 ppm. Elemental Analysis (%) calcd. for C17H37Al2Nd (439.68): C 46.44, H 8.48; found: C 46.52, H 8.09.
6.7 [(C5Me4H)La(AlMe4)2]2 (2e)
Following the procedure described above, [La(AlMe4)3] (1e) (201 mg, 0.50 mmol) and H(C5Me4H) (65 mg, 0.53 mmol) yielded 2e (191 mg, 0.22 mmol, 88%) as colorless crystals. 1H NMR (500 MHz, [D6]benzene): δ = 5.62 (s, 2H, CH), 1.87 (s, 12H, Me), 1.75 (s, 12H, Me), −0.27 (s, 48H, AlMe4) ppm. 13C NMR (126 MHz, [D6]benzene): δ = 126.1, 126.0, 117.0, 13.5, 11.3, 2.3 (br. s, AlMe4) ppm. Elemental analysis (%) calcd. for C34H74Al4La2 (868.71): C 47.0, H 8.6; found: C 47.1, H 8.2.
6.8 General procedure for the preparation of cations
To a solution of [{(Cp′)La(AlMe4)2}2] in [D6]benzene a solution or suspension of the cocatalyst (B(C6F5)3, [Ph3C][B(C6F5)4], [PhNMe2H][B(C6F5)4],) in [D6]benzene was added. The solution was transferred to a Teflon-valved type NMR tube.
6.9 [{(Cp′)La(AlMe4)2}2] with [Ph3C][B(C6F5)4] (3a)
Following the procedure described above [{(Cp′)La(AlMe4)2}2] (10 mg, 0.02 mmol) with [Ph3C][B(C6F5)4] (21 mg, 0.02 mmol) gave a clear faint yellow solution after several minutes of mixing. 1H NMR (500 MHz; [D6]benzene): δ = 7.15-7.02 (m, 15H, Ph3CMe), 5.38 (s, 1H, CH), 2.03 (s, 3H, Ph3CMe), 1.63 (s, 6H, CpMe), 1.54 (s, 6H, CpMe), −0.36 (br. s, 9H, AlMe3), −0.53 (br. s, 12H, AlMe4) ppm. 13C{1H} NMR (126 MHz, [D6]benzene): δ = 150.0 (C6F5), 149.5 (Ph), 148.1 (C6F5), 144.4, 139.9, 138.2, 137.8, 136.3 (C6F5), 135.7, 126.2 (CpMe), 120.0, 52.8 (Ph), 30.7 (Ph3CMe), 12.6, 10.8 (CpMe), 3.5 (AlMe4), −7.1 (AlMe3) ppm. 11B{1H} NMR (161 MHz, [D6]benzene): δ = −16.2 ppm. 27Al NMR (130 MHz, [D6]benzene): δ = 159 ppm. 19F NMR (471 MHz, [D6]benzene): δ = −131.2 (d, o-F), −160.3 (t, p-F), −165.0 (t, m-F) ppm.
6.10 [{(Cp′)La(AlMe4)2}2] with [PhNMe2H][B(C6F5)4] (3b)
Following the procedure described above [{(Cp′)La(AlMe4)2}2] (11 mg, 0.03 mmol) with [PhNMe2H][B(C6F5)4] (21 mg, 0.03 mmol) gave a clear faint yellow solution. 1H NMR (500 MHz; [D6]benzene): δ = 7.01-6.98 (m, 2H, Ph), 6.88-6.82 (m, 3H, Ph), 5.46 (s, 1H, CH), 2.24 (s, 6H, NMe2), 1.70 (s, 6H, Me), 1.62 (s, 6H, Me), −0.50 (br. s, 12H, AlMe4), −0.65 (br. s, 9H, AlMe3) ppm. 13C{1H} NMR (126 MHz, [D6]benzene): δ = 150.0, 148.1, 147.5, 139.9, 138.2, 137.9, 136.3 (C6F5), 126.4 (CpMe), 124.3, 120.3 (Ph), 119.8, 45.4 (PhNMe2), 12.7 (CpMe), 10.8 (CpMe), 1.2 (AlMe4), −6.1 (AlMe3) ppm. 11B{1H} NMR (161 MHz, [D6]benzene): δ = −16.3 ppm. 27Al NMR (130 MHz, [D6]benzene): δ = 199 ppm. 19F NMR (471 MHz, [D6]benzene): δ = −131.3 (d, o-F), −160.4 (t, p-F), −164.8 (t, m-F) ppm.
6.11 [{(Cp′)La(AlMe4)2}2] with B(C6F5)3 (4)
Following the procedure described above [{(Cp′)La(AlMe4)2}2] (11 mg, 0.02 mmol) with B(C6F5)3 (12 mg, 0.02 mmol) gave a clear colorless solution (Caution: concentrated solutions and the solid reaction product of this reaction are highly shock sensitive). 1H NMR (500 MHz, [D6]benzene): δ = 5.63 (s, 2H, CH), 1.78 (s, 12H, Me), 1.67 (s, 12H, Me), 0.72 (s, BMe3), −0.04 (br. s, 12H, (C6F5)2Al(μ-Me)2 and (C6F5)Al(μ-Me)2Me), −0.17 (br. s, 6H, (C6F5)Al(μ-Me)2Me) ppm. 13C{1H} (126 MHz, [D6]benzene): δ = 150.8, 149.1, 144.5, 143.1, 142.3, 141.1, 139.2, 138.3, 136.5, 130.3 (C6F5), 117.9, 117.5 (CpCH3), 15.1 (CpCH3), 14.9 (B(CH3)3), 13.5, 12.9, 12.2, 11.4, 10.1 (CpCH3), 4.7 (s br, Al(CH3)3), 4.0 (br. s, Al(CH3)3), −8.1 (br. s, (C6F5)2Al(CH3)2), −8.5 (br. s, (C6F5)2Al(CH3)2) ppm. 11B{1H} (161 MHz, [D6]benzene): δ = 86.3 (s, BMe3) ppm. 27Al NMR (130 MHz, [D6]benzene): δ = 158 (br. s, AlMe3), 145 (br. s, Al(C6F5)2Me2) ppm. 19F NMR (471 MHz, [D6]benzene): δ = −122.0 (br. s, 4F, o-F), −123.5 (d, 1F, o-F), −124.1 (d, 1F, o-F), −150.2 (t, 1F, p-F), −153.9 (br. s, 2F, p-F), −160.0 (br. s, 4F, m-F), −162.8 (t, 2F, m-F) ppm.
6.12 General procedure for polymerization of isoprene
A detailed polymerization procedure (run 3, Table 1) is described as a typical example. To a solution of 2e (9 mg, 0.02 mmol) in toluene (8 mL) 1 equiv of B(C6F5)3 (10 mg, 0.02 mmol) was added and the mixture aged at 40 °C for 20 min. After the addition of isoprene (2.0 mL, 20 mmol) the polymerization was carried out at 40 °C for 24 h. The polymerization mixture was poured onto a large quantity of acidified isopropanol containing 0.1% (w/w) 2,6-di-tert.butyl-4-methylphenol as a stabilizer. The polymer was washed with isopropanol and dried under vacuum at ambient temperature to constant weight. The polymer yield was determined gravimetrically.
6.13 Single crystal X-ray structures
Crystal data and details of the structure determination are presented in Table 2. Single crystals were placed in a nylon loop containing Paratone-N oil (Hampton Research) under argon atmosphere, then mounted directly into the N2 cold stream (Oxford Cryosystems Series 700) on a Bruker AXS SMART 2K CCD diffractometer. Data were collected by means of 0.3° ω-scans in four orthogonal φ-settings using MoKα radiation (λ = 0.71073 Å). Data collection was controlled using the program SMART [23], data integration using SAINT [24], and structure solution and model refinement using SHELXS-97 and SHELXL-97 [24].
Crystallographic data for compounds 2c, 2d, and 2e.
| 2c | 2d | 2e | |
| Formula | C17H37Al2Sm | C17H37Al2Nd | C34H74Al4La2 |
| Fw | 445.78 | 439.67 | 868.67 |
| Color/habit | Brown-red/plate | Blue/prism | Colorless/prism |
| Crystal dimensions [mm3] | 0.43 × 0.35 × 0.08 | 0.52 × 0.28 × 0.22 | 0.52 × 0.27 × 0.23 |
| Cryst syst | Monoclinic | Monoclinic | Triclinic |
| Space group | P21/c | P21/c | P-1 |
| a [Å] | 11.8277(4) | 11.8968(3) | 10.4725(8) |
| b [Å] | 7.4655(3) | 7.4738(2) | 10.7491(8) |
| c [Å] | 24.1851(9) | 24.2583(7) | 11.9506(9) |
| α [°] | 90 | 90 | 74.043(1) |
| β [°] | 97.166(1) | 97.199(1) | 66.316(1) |
| γ [°] | 90 | 90 | 63.9320(1) |
| V [Å3] | 2118.86 | 2139.91(10) | 1098.70(14) |
| Z | 4 | 4 | 1 |
| T [K] | 100(2) | 123(2) | 123(2) |
| ρcalcd [mg m−3] | 1.397 | 1.365 | 1.313 |
| μ [mm−1] | 2.846 | 2.500 | 2.016 |
| F(000) | 908 | 900 | 444 |
| θ range [°] | 1.74/30.11 | 1.69/30.14 | 1.87/30.11 |
| Index ranges | −16 ≤ h ≤ 16, −10 ≤ k ≤ 10, −34 ≤ l ≤ 34 | −16 ≤ h ≤ 16, −10 ≤ k ≤ 10, −34 ≤ l ≤ 34 | −14 ≤ h ≤ 14, −15 ≤ k ≤ 15, −16 ≤ l ≤ 16 |
| No. of rflns integrated | 24336 | 35103 | 18646 |
| No. of indep rflns/Rint | 6225/0.0209 | 6307/0.0175 | 6470/0.0132 |
| No. of obsd rflns (I > 2σ(I)) | 6015 | 6129 | 6330 |
| Data/params/restraints | 6225/242/30 | 6307/241/30 | 6470/242/30 |
| (I > 2σ(I))a | 0.0165/0.0403 | 0.0137/0.0352 | 0.0126/0.0331 |
| (all data)a | 0.0175/0.0407 | 0.0142/0.0354 | 0.0130/0.0333 |
| GOF (on F2)a | 1.192 | 1.177 | 1.120 |
| Largest diff peak and hole [e Å−3] | 0.565/−0.855 | 0.366/−0.616 | 0.563/−0.249 |
All data sets were subject to absorption correction using multi-abs methodology [25].
Non-coordinating methyl groups were refined as rigid and rotating (difference Fourier density optimization) around the respective Al–C bonds. Coordinating methyl groups were refined as rigid pyramidal groups with the same C–H and H---H distances as for the previous, but with the threefold axis of the pyramidal rigid group allowed to be non-parallel with the C-Al bond axis. The isotropic displacement parameters for all methyl H-atoms were set to be 1.5 times that of the pivot C-atom.
CCDC deposition numbers CCDC 757975 - 757977 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre, via http://www.ccdc.cam.ac.uk/data_request/cif.
Acknowledgment
Financial support from the Norwegian Research Council (Project No. 182547/I30) is gratefully acknowledged.
1 Noteworthy, catalyst mixtures derived from [(C5Me4SiMe3)-a(AlMe4)2] and [Ph3C][B(C6F5)4] (A) were recently reported to benactive in the polymerization of butadiene at−40 °C [17a].