

1 Introduction
There is fundamental interest in reactivity of organolanthanide complexes with unsaturated organic small molecules, because this is the source for developing new catalytic reactions and catalysts [1]. Furthermore, insertion of an organic functional group into a metal−ligand bond represents one of the most useful processes for the formation of carbon−carbon and carbon−heteroatom bonds and plays a key role for many organolanthanide reactions and catalysts [2]. For example, olefin insertion into a metal−hydrogen (carbon or heteroatom) bond is a product-determining step in the lanthanocene-catalyzed cyclization/functionalization reactions [3]. So the studies on insertions of organolanthanide complexes with unsaturated organic small molecules and their applications in organic synthesis have experienced extremely important development and display many novel structures of the intermediate compounds and reactivity patterns during the last two decades [4–7].
Organolanthanide amides are one of the most important organolanthanide derivatives due to the balance between the stability and activity of the lanthanide−nitrogen bond [8]. They not only take part in many organometallic reactions, but also are the real catalysts in organolanthanide-catalyzed intramolecular hydroamination/cyclization reactions [9,10]. This contribution is intended to make a review of the developments in reaction chemistry of organolanthanide complexes with the N−H bonds toward some unsaturated organic small molecules, that is mainly based on some recent research results in our group. Some related results reported by other groups are also included. To avoid the overlap with organolanthanide-catalyzed hydroaminations reviewed by Hong and Marks [11], the unsaturated organic functional groups in this article are mainly limited to the carbon−heteroatom double and triple bonds.
2 The formal N−H additions of the Ln-bonded NHR anion ligands
The first example of organolanthanide-mediated N−H bond addition with carbodiimide was reported in 2004 [12]. The lanthanocene primary amides [(C5H5)2LnNHR]2 react with iPrN=C=NiPr to form the corresponding formal N−H bond addition products (C5H5)2Ln[RNC(NHiPr)NiPr] (R = tBu, Ln = Yb(1a), Er(1b), Dy(1c), Y(1d); R = Ph, Ln = Yb(2)), providing a potential protocol to synthesize organolanthanide derivatives with asymmetrical guanidinate ligands (Scheme 1). This result is significantly different from the observations of the insertion of carbodiimide into the Ln−N bond of organolanthanide secondary amides, which usually forms the symmetrical coordinated guanidinate complexes [13].
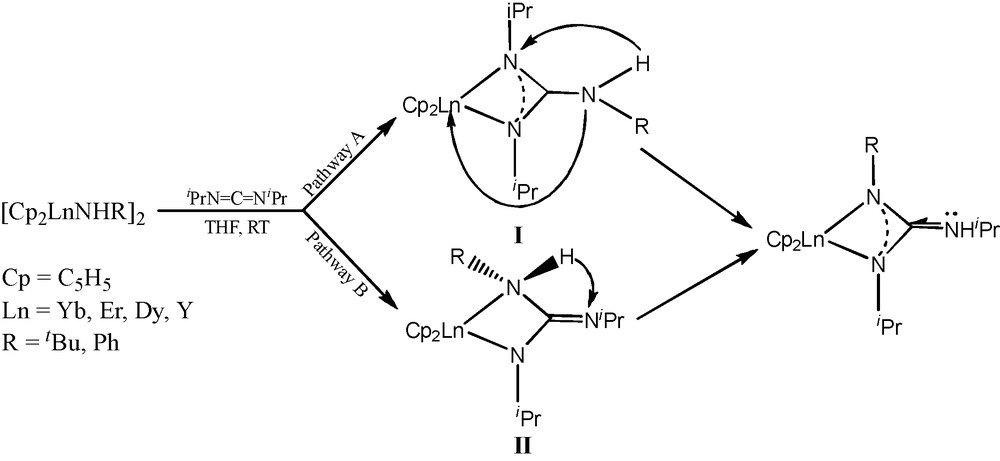
As shown in Scheme 1, there are two possible pathways for the formation of these complexes. Pathway A involves 1,3-migration of the NHR group to the carbon atom of coordinated carbodiimide forming the intermediate I, analogous to the reactivity of lanthanocene secondary amido derivatives with carbodiimide, followed by tautomerization to the final products. The alternative pathway B involves intramolecular coupling, followed by 1,3-hydrogen migration.
Given the expectation that the N−H addition could be distinguished from the Ln−N addition if the presence of chelating substituents could prevent from migration the amido in an effective carbodiimide reaction process, a series of lanthanocene complexes featuring different functionalized primary amido ligands were synthesized (Scheme 2), and their reactivities toward carbodiimides were investigated [14].
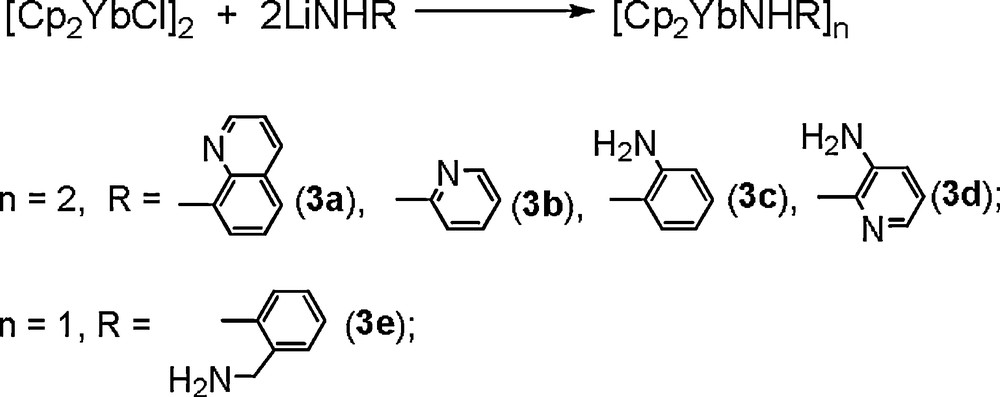
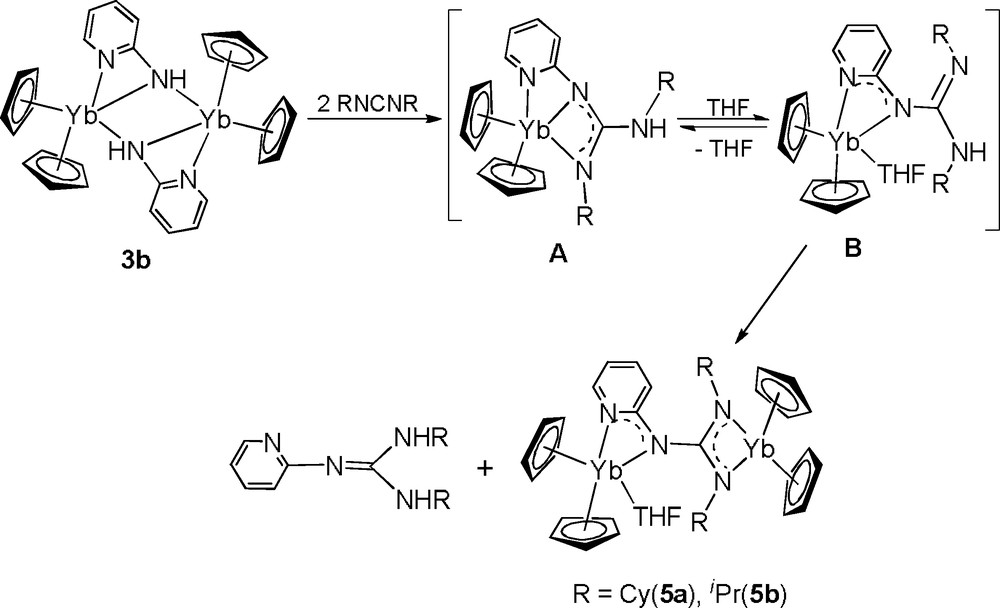
Similar to the results observed in reaction of [(C5H5)2LnNHR1]2 (R1 = tBu, Ph) with iPrN=C=NiPr, 3a reacts with two equivalents of RN=C=NR (R = iPr, Cy) in THF to give the products from formal carbodiimide insertion into the N−H bond, 4a and 4b (Scheme 3). However, 3b reacts with two equivalents of RN=C=NR under the same conditions to give the dianionic guanidinate complexes 5a and 5b in moderate yield. 5a and 5b may result from the carbodiimide insertion into the N−H bond of 3b [12], followed by the intermolecular guanidine elimination (Scheme 4). The driving force for the partial guanidine elimination reaction of the putative guanidinate intermediates probably results from the acidity of the N−H proton and the chelating effect of the pyridyl group that seems to favor higher coordination numbers than the corresponding phenyl-substituted guanidinate [12]. The additional pyridyl substituent should help to stabilize the bridging coordination of dianionic guanidinate to two Yb3+ centers.

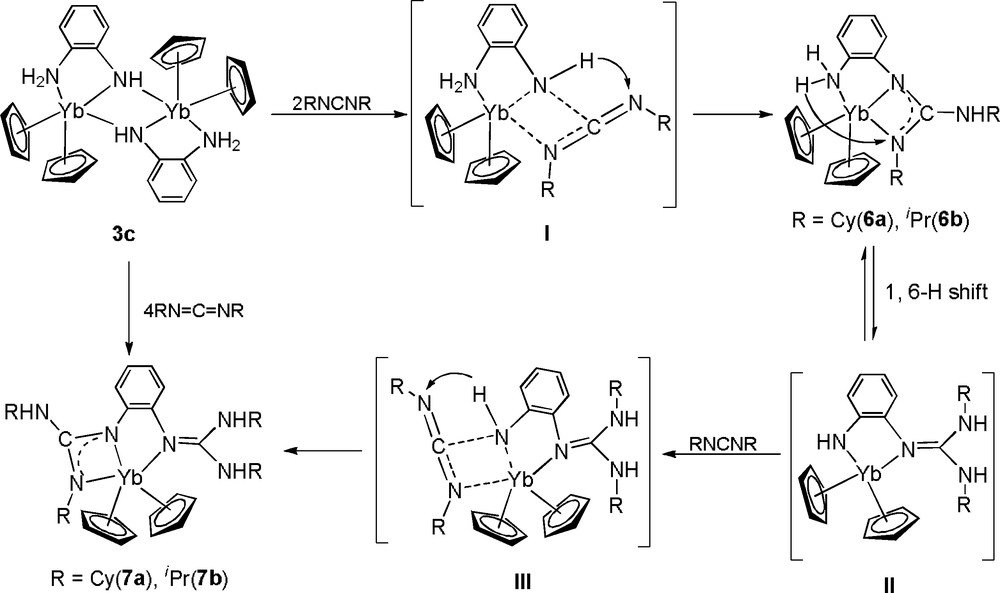
The reaction of 3c with two equivalents of CyN=C=NCy in THF at room temperature leads to the isolation of the single N−H addition product 6a in 65% yield, while treatment of 3c with four equivalents of CyN=C=NCy gives the double N−H addition product 7a in 58% yield. Similarly, 6b and 7b can be respectively obtained by controlling the stoichiometric ratio of 3c and iPrN=C=NiPr. It is found that complex 7 could also be obtained by reacting 6 with one equivalent of RN=C=NR. The formation of compounds 7 represents a rare example of multiple N−H activation of organolanthanide complexes.
A possible reaction pathway for the formation of 6 and 7 is proposed in Scheme 5. In the initial step, coordination of carbodiimide and nucleophilic attack of the amido at the carbodiimide central carbon atom would yield the intermediates I [15]. Then, the 1,3-hydrogen migration from the amido to the more basic uncoordinated nitrogen affords the formal single N-H bond addition products 6. Furthermore, an intraligand proton transfer from the chelating NH2 group to the metal-bound guanidinate group [16] and subsequent formal insertion of a second carbodiimide molecule into the N−H bond of the newly formed amido group yields the double N−H addition products 7.
3d reacts with CyN=C=NCy in 1:4 ratio to form the double-addition product 8 in a good yield with complete chemoselectivity. A pathway similar to that proposed for the formation of 7 is very likely to account for the formation of 8. Namely, the first step in this multiple N−H bond activation system is a carbodiimide molecule insertion into the N−H bond of the amido of 3d, giving i as a transient intermediate. i can adopt an ii structure with the aid of additional intramolecular hydrogen bonding. As soon as iii is formed by intramolecular proton transfer, it can then insert into a second carbodiimide molecule to form iv. Last, the converse 1,8-H shift gives the more stable complex 8.
Complex 6 is unstable to heat and is readily transformed to the unusual neutral guanidine-substituted guanidinate dianion lanthanide complex 9 under refluxing in THF with intermolecular elimination of neutral diguanidine, as indicated in Scheme 6. These results demonstrate that the chelating effect of pyridyl rings can impart to the guanidinate complexes a unique reactivity and initiates the unexpected reaction sequence.

However, only the monoaddition products 10a and 10b are obtained, when 3e is treated with one equivalent of RN=C=NR in THF (Scheme 7). Attempts to isolate the di-insertion products similar to 7 are unsuccessful, even when a large excess of carbodiimide is added under forcing conditions. In marked contrast to 6, the amino group of 10a and 10b is inert and undergoes neither the direct addition to excess RN=C=NR (R = Cy, iPr) that occurs directly at the free NH2 group nor the proton transfer to the newly formed guanidinate ligand under the conditions involved. This may be attributed to both its lower nucleophilicity caused by the chelating coordination and inherent weak acidity. These results are consistent with the relative acidity of aromatic amine greater than aliphatic amine and thus also provide evidence to support the mechanistic consideration that the proton transfer from the coordinated amino group to the newly formed guanidinate group is the driving force for the further hydroamination reaction of the monoaddition intermediate with carbodiimide in the formation of 7 and 8. Since aliphatic NH2 is not acidic enough to protonolyze the aryl-substituted guanidinate ligand, reforming an amido group, the further guanylation for 10 is prevented.

As shown in Scheme 8 [17], 11, prepared by the protonolysis of Cp3Yb with 2,6-diaminopyridine (2,6-(H2N)2C5H3N), reacts with two equivalents of iPrN=C=NiPr in THF at 0 °C to give 12, indicating that carbodiimide inserts into each the N–H bond of two amido groups of 11. When the THF solution of 12 is kept at room temperature for two weeks, it is slowly converted into the unusual guanidine-substituted dianionic guanidinate complex 13 via an unprecedented disproportionation of the diguanidinate ligand. Furthermore, it is found that under the heating condition (about 70 °C), 13 can undergo the partial cyclopentadiene elimination to give a rare linear tetrametallic complex 14. 14 represents the first example of linked diguanidinate trianion complex. These results show that the intramolecular proton transfer reaction can take place not only between guanidinate groups but also between the guanidine substituent and cyclopentadienyl co-ligand.

To explore the generality of isomerization of pyridyldiguanidinate ligands, we further examined the reaction of related (pyridyldiamido-2,3)ytterbium complex [Cp2Yb(THF)]2[μ-(HN)2(C5H3N-2,3)] (15) with CyN=C=NCy [18]. In contrast to the observation for 11, treatment of 15 with CyN=C=NCy gives only the addition/isomerization product 16 (Scheme 9). These differences may be attributed to the weaker acidity of the guanidine group at the 3-position as compared with that at the 6-position, which enhances the tendency of the proton transfer from guanidinate at the 2-position to guanidinate at 3-position. These observations imply that subtle differences in the position of guanidinate groups on the pyridyl ring would bring about differences in their reactivity.
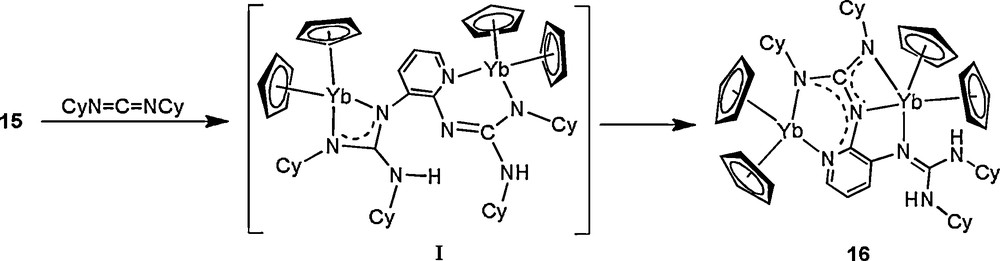
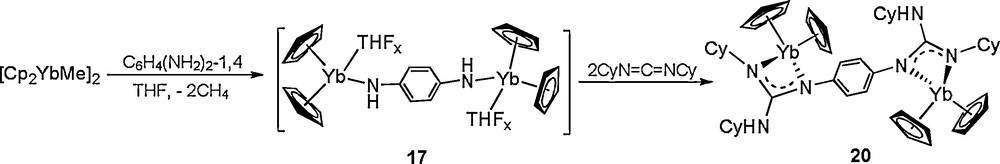
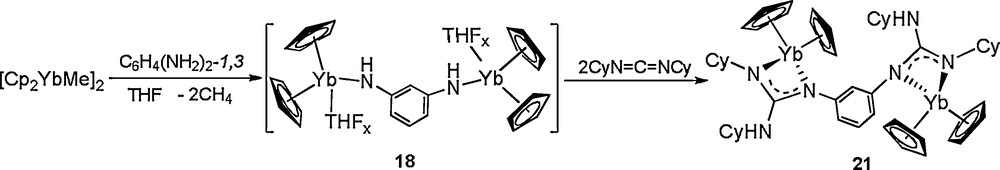
To find out the factors that affect the proton transfer, several novel aryl diamido complexes 17, 18, and 19, are synthesized in situ by treatment of [Cp2YbMe]2 with the corresponding aryldiamine, and their doubly N−H activation properties are also probed by exploring their reactivity toward carbodiimides. Treatment of complexes 17-19 with an excess of CyN=C=NCy, even under prolonged heating at 110 °C, affords only the products of formal double CyN=C=NCy insertion into the N−H bond of the Yb-bonded NHAr group, to yield complexes 20, 21, and 22, respectively (Schemes 10–12). These results indeed demonstrate that the reactivity behavior of the linked diamido ligand strongly depends on the nature of the bridge system. The pyridyldiamido ligand bonded to the lanthanide metal can undergo a clean tandem diguanylation/isomerization to afford a variety of rare mixed neutral guanidine/dianionic guanidinate complexes, while the resultant aryldiguanidinate ligands are stable under the same conditions.

It is known that addition of amine to carbodiimides is a straightforward and atom-economical route to synthesize neutral organic guanidines. Noticeably, such catalytic reactions have recently been realized by using organolanthanide complexes as a catalyst precursor [19]. The half-sandwich rare earth metal alkyl complexes can promote the addition of a second amine or primary aromatic amine with carbodiimide to give the corresponding neutral guanidinates in high yields. The corresponding rare-earth metal guanidinate complexes have been confirmed to be true catalyst species in this process (Schemes 13 and 14) [19b].


The reactions of organolanthanide complexes containing the N−H bonds with other unsaturated organic small molecules such as isocyanate, isothiocyanate, nitrile, and isocyanide have also been studied [20]. The organolanthanide complexes with the benzophenon hydrazonido(1−) ligand, [Cp2Yb(μ-NHN=CPh2)]2 (23) and Cp2Er[η2-NHN=CPh2](THF) (24), are synthesized by abstraction of cyclopentadienyl from Cp3Ln with benzophenon hydrazone. They react with phenyl isocyanate to give the formal N−H bond addition products Cp2Ln(η2-OC(NHPh)NN=CPh2)(HMPA) (Ln = Yb(25), Er(26)). However, they react with PhNCS to give the Ln−N insertion products Cp2Ln(η2-SC(NHN=CPh2)NPh)(HMPA) (Ln = Yb(27), Er(28)) under the same conditions (Scheme 15).

Further investigation results indicate that PhCN readily inserts into the Er−N bond of 24 with 1,3-hydrogen migration to form the organolanthanide heterometallacycles Cp2Er[η2-NHC(Ph)NN=CPh2] (29), similar to the observations of the reactions of scandium hydrazido complexes with acetonitrile [21]. However, tBuNC does not insert into the Er−N bond of 24 under the same conditions and treatment of 24 with tBuNC leads to the isolation of the solvent-free dimer [Cp2Er(μ-η1:η2-NHN=CPh2)]2 (24’) (Scheme 16).

The organolanthanide-induced isocyanate diinsertion into the N−H bond has also been studied and provides an alternative, versatile approach to the construction of diureido ligands. Interestingly, the further investigation results indicated that the diinsertion is reversible, a second isocyanate can exchange with the inserted isocyanate unit from the diinsertion products to yield another diinsertion derivative [22]. 3b reacts with 4-nitrophenyl isocyanate to give the isocyanate diinsertion products 30a and 30b regardless of the equivalency of the isocyanate reagent employed. The more bulky isocyanate (2,6-iPr2C6H3NCO) also undergoes double insertion into 3b, affording 31a and 31b (Scheme 17). The presence of electron-withdrawing and electron-donating substituents on the pyridyl ring does not appear to alter the product selectivity. For example, the diinsertion of phenyl isocyanate into substituted pyridyl complexes [Cp2LnNHPyMe]2 (PyMe = 4-methyl-2-pyridyl) (33) and [Cp2LnNHPyCl]2 (PyCl = 5-coloro-2-pyridyl) (34) leads to the formation of 35a-b, and 36a-c, respectively (Scheme 18).


The monoinsertion intermediate 37·HMPA can be trapped during this diinsertion process by adding HMPA. Interestingly, the mixed diinsertion complex Cp2Yb[η2:η1-PyNCON(C6H3iPr2-2,6)CONHPh] (38) can be prepared by allowing the 2,6-iPr2C6H3NCO to react first with PyNH2 and then with the Cp3Yb followed by inserting with PhNCO or by the reaction of 37 with PhNCO (Scheme 19).

A mechanism involving a direct addition of isocyanates to the N−H bonds of 3b and the initially formed monoinsertion intermediate, presumably via a polar four-center σ-bond metathesis transition state is shown in Scheme 20.

As shown in Scheme 21, 31a reacts with excess phenyl isocyanate to yield the unexpected replacement of 2,6-iPr2C6H3NCO units inserted into the N–H bond by PhNCO molecules, wherein the newly formed ligand by exchanging 2,6-iPr2C6H3NCO with PhNCO has been structurally characterized in their protonated form PyNHCON(Ph)CONHPh (39) and PyNHCON(C6H3iPr2-2,6)CONHC6H3iPr2-2,6 (40).

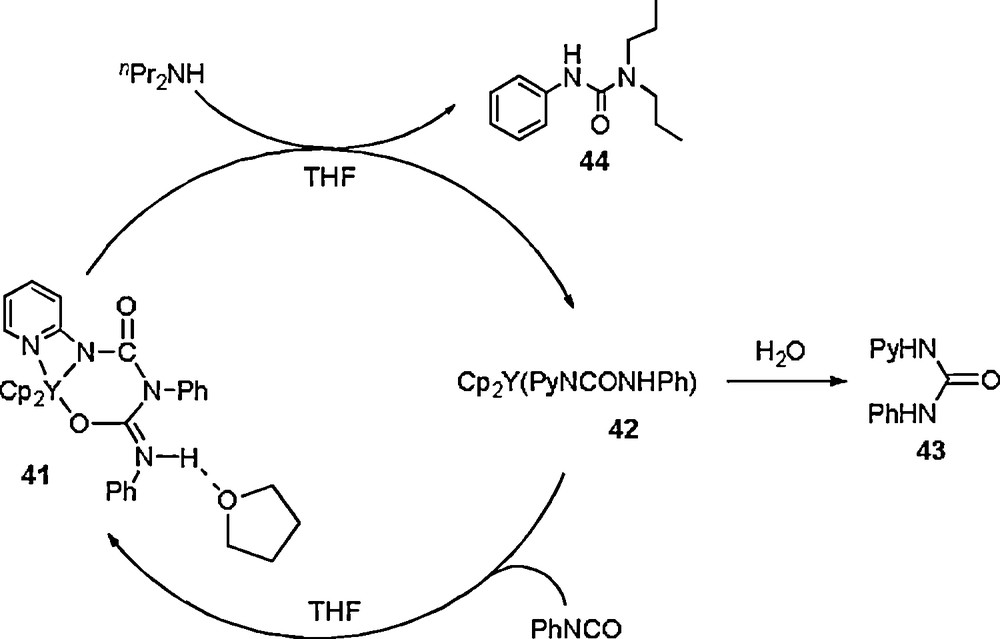
It is found that treatment of 41 with excess nPr2NH leads to the abstraction of one PhNCO unit by amine, giving Cp2Y[OC(NHPh)NPy] (42) and nPr2NC(O)NHPh (43) in 96% and 41% yield, respectively, indicating that the second insertion reaction is reversible under certain conditions (Scheme 22) [23].
Reaction of Cp2LnNiPr2(THF) with two equivalents of anthranilonitrile in THF at room temperature affords the isolated and structurally characterized complexes Cp2Ln[κ3-(4-NH(C8N2H4)(2-NH2C6H4)] (Ln = Er(45), Y(46)) in moderate or high yields [24]. Structural analysis reveals that an intermolecular nucleophilic addition/cyclization of anthranilonitrile takes place accompanied by elimination of HNiPr2 to form a novel [(4-NH(C8N2H4)(2-NH2C6H4)]- ligand. These compounds can also be obtained from the reaction of Cp2LnNiPr2(THF) with one equivalent of anthranilonitrile under the same conditions in lower yields.
A proposed mechanism for the formation of 45 and 46 is illustrated in Scheme 23. The first step is protonolysis of the diisopropylamine ligand by an anthranilonitrile molecule to generate the intermediate I. Then the nitrile group from another anthranilonitrile molecule inserts into the Ln−N bond of I, giving A. The following 1,3-hydrogen shift leads to the formation of B. The final products are formed by nitrile insertion followed by 1,3-hydrogen shift again.

Reaction of Cp2ErNiPr2(THF) with anthranilonitrile and iPrN=C=NiPr in 1:1:1 ratio gives two isolated products [Cp2Er(μ-κ1:κ2-NCC6H4N(H)C(NHiPr)=NiPr)]2 (47) and 45 in 47% and 9% yields, respectively. This result maybe indicates that nucleophilic additions of carbodiimide and anthranilonitrile with I are competitive. The proposed mechanism for the formation of 45 is shown in Scheme 24, a typical process of the σ-bond metathesis and further insertion of carbodiimide molecules, companying with 1,3-hydrogen shift [4e].

Significantly, reaction of Cp2LnCl(THF) with two equivalents of [2-NCC6H4HNLi(THF)]n (48) in THF at room temperature affords the 4-iminoquinazolinate complexes Cp2Ln[κ3-(4-NH=(C8N2H4)(2-NHC6H4)]Li(THF)3 (Ln = Er(49), Y(50)) in high yields, as shown in Scheme 25 [25]. Further investigations indicate that reaction of Cp2LnCl(THF) with one equivalent of 48 under the same conditions also gives 49 and 50, albeit in low yields. However, the dimerization/cyclization of 48 cannot occur during the preparation of 48. These results show that the activation of the adjacent nitrile group via the coordination interaction with the Ln3+ ions plays a key role to this nucleophilic addition.

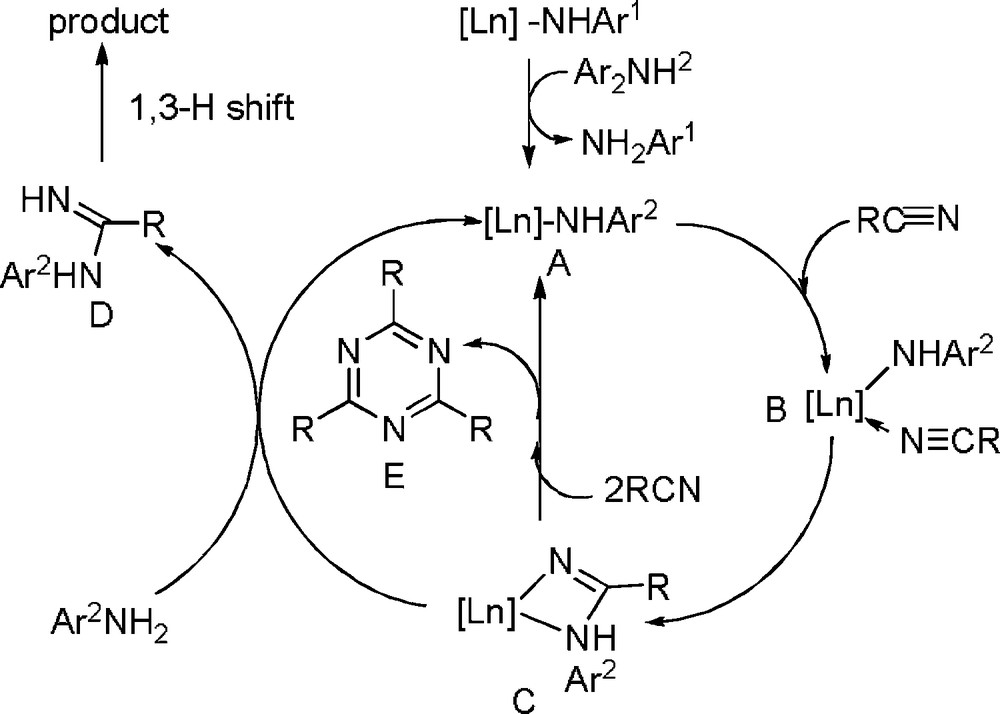
Recently, Shen et al. reported a one-step synthesis of monosubstituted N-arylamidinates via addition of amines to nitriles catalyzed by ytterbium amides [26]. The reactions of aromatic nitriles with aromatic primary amines proceed very well to give excellent yields with 5 mol % ytterbium at 100 °C under solvent-free conditions (Scheme 26), which provides a compensative method for the published intramolecular nucleophilic addition of amine to activated nitrile methods with Ln(OTf)3 and/or SmI2 as the catalysts [27], which frequently suffered from difficulty in synthesis of monosubstituted amidines. However, the reaction with aliphatic amines has not yet been successful.

A proposed mechanism for the reaction of nitriles with amines is shown in Scheme 27. Reaction of an aniline with the lanthanide amide gives the new amido intermediate A through an acid-base reaction [11]. A nitrile is then coordinated to the center metal forming a complex B. An intramolecular insertion of amide to cyano of nitrile gives the corresponding intermediate C as reported previously [28]. Intermediate C undergoes protonation by amine to release the product and to generate the lanthanide amide active species A. When the reaction of intermediate C with additional nitrile is more favorable than that with amine, triazine E is produced as the main product (Scheme 27).
3 Activity of the Ln-bonded neutral NH2 groups
In 2005, we reported the first example of the addition of the amino group to phenyl isocyanate [29]. Two organolanthanide complexes with the bisfunctional ligands [Cp2Yb(o-H2NC6H4S)]2·2THF (51) and Cp2Yb(p-H2NC6H4S)(THF) (52) are facilely prepared by the protonolysis of Cp3Yb with the corresponding amino-substituted thiophenol. 51 reacts with two equivalents of PhNCO in THF at ambient temperature to give an unexpected benzothiazole-2-oxide complex Cp2Yb(μ-η1, η3-OSNC7H4)]2 (53) in 53% yield, indicating that the adjacent NH2 group undergoes immediately a tandem intramolecular hydroamination/PhNH2 elimination on the insertion of PhNCO into the Yb−S bond (Scheme 28). However, treatment of 52 with two equivalents of PhNCO under the same conditions provides only the dinuclear insertion product {Cp2Yb[μ-η1:η3-OC(p-H2NC6H4S)NPh]}2·2THF (54) (Scheme 29). This result provides that PhNCO is preferentially added to the Yb−S bond rather than the N−H bond. The free NH2 group at para-position is inert, and undergoes neither the competitive addition to PhNCO nor the further addition to the newly formed OC(p-H2NC6H4S)NPh ligand under the conditions involved.


A possible reaction pathway for the formation of 52 is proposed in Scheme 28. Coordination of PhNCO and migration of o-H2NC6H4S give the insertion intermediate (I). A sequential addition of the activated N−H to the C=N double bond results in cyclization via a four-centered σ bond metathesis process. Reductive elimination of PhNH2 generates the thiazolate ring.
The reactivity of complexes 51 and 52 with carbodiimides have also been studied [30–32]. Addition of two equivalents of iPrN=C=NiPr to the THF solution of 51 at ambient temperature results in a direct N–H bonds addition product [Cp(THF)Yb(μ-η3:η1-SC6H4N=C(NHiPr)NiPr)]2 (55), indicating that the adjacent NH2 group undergoes immediately a tandem intermolecular addition/1,3-hydrogen shift/CpH elimination, to yield a novel dianionic guanidinate ligand [SC6H4N=C(NHiPr)NiPr]2-, as shown in Scheme 30 [30].
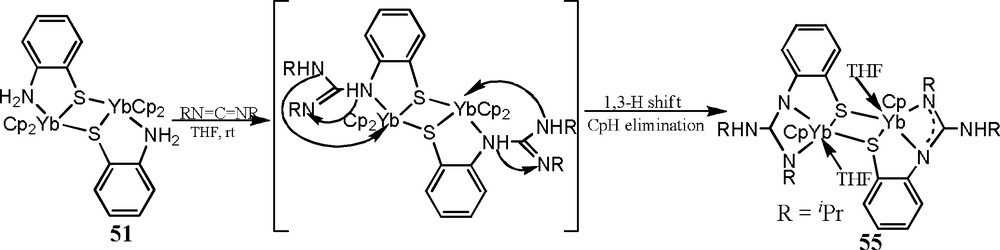
In contrast of the reaction of 51 with the phenyl isocyanate, only the addition of the activated N–H bonds of the NH2 group to the C=N bonds of carbodiimide molecule occurs, the Ln–S σ-bond is inert to the insertion of carbodiimide. This might be attributed to the differences of the activity between PhNCO and iPrN=C=NiPr [13].
To further investigate the selectivity of the addition reaction, we also synthesized the complex [Cp2Dy(o-H2NC6H4S)]2·3THF (56) by the reaction of Cp3Dy with o-aminothiophenol in THF at room temperature, and examined its reactivity toward N, N’-diisopropylcarbodiimide. X-ray diffraction analysis result reveals an unsymmetrical intermolecular N–H···O hydrogen bonding-mode in 56, wherein one NH2 group interacts with two THF molecules, another amino group only interacts with one THF molecule. Reaction of 56 with two or one equivalent of iPrN=C=NiPr in THF at room temperature gives the partial amino group addition product 57, as shown in Scheme 31, indicating that only one amino group adds with the carbodiimide molecule, another amino group is retained. The residual NH2 group also interacts with one THF molecule via the intermolecular N–H···O hydrogen bond, as confirmed by X-ray diffraction analysis. To identify the role of the intermolecular hydrogen bond interaction, the reaction is carried out in toluene. When Cp3Dy reacts with o-aminothiophenol in toluene at room temperature, and subsequently with iPrN=C=NiPr, complex 57 and small amount of [Cp2Dy(o-H2NC6H4S)]2·2THF (58) can be isolated (recrystallization in the mixed solution of THF and toluene). 58 is a symmetric dimer geometry with two intermolecular hydrogen bond interactions. After further crystallization of the above mother liquor at low temperature, trace amounts of purple crystals [Cp(THF)Dy(μ-η3:η1-SC6H4N=C(NHiPr)NiPr)]2 (59), which is a amino group completed addition product, are obtained. These results maybe indicate that the partial addition is mainly affected by the character/radii of the rare earth metal Dy3+ions, not the intermolecular hydrogen bond interactions.
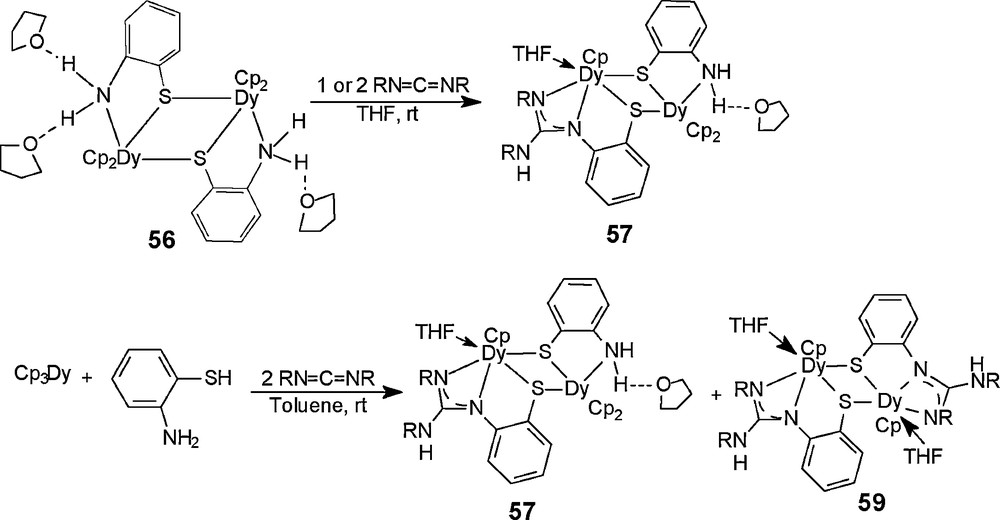
It should be noted that the reaction of 56 with two equivalents of iPrN=C=NiPr in THF under the reflux temperature also gave 57, the residue NH2 group do not continue to add to the C=N bonds of another carbodiimide molecule. To obtain the partial amino group addition product similar to 57, the reaction of 51 with 55 in a 1:1 ratio has also been investigated. However, this reaction does not take place, and the original materials are recovered.
To extend the scope of the addition, reactions of CpEr(o-H2NC6H4S)2 with one or two equivalents of iPrN=C=NiPr have also been studied. As shown in Scheme 32, reaction of Cp3Er with two equivalents of o-aminothiophenol in THF at room temperature, and subsequently with two equivalents of iPrN=C=NiPr yield the bis-addition product (C5H5)Er[SC6H4NC(NHiPr)2]2 (60). Both of the two NH2 groups add to the C=N bonds of carbodiimide molecules, but no abstraction of cyclopentadienyl takes place. This may be attributed to the steric hindrance of two guanidinate ligands [SC6H4NC(NHiPr)2]-. However, when Cp3Er reacts with two equivalents of o-aminothiophenol, and subsequently with one equivalent of iPrN=C=NiPr, an organic disulfide (iPrHN)2CNC6H4SSC6H4NC(NHiPr)2 (61) has been obtained; the residual solids are a mixture containing the metal ion Er3+.
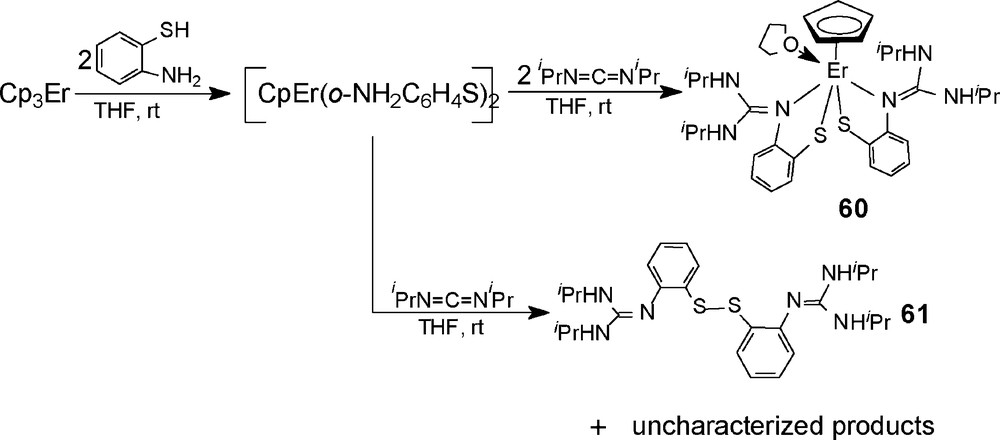
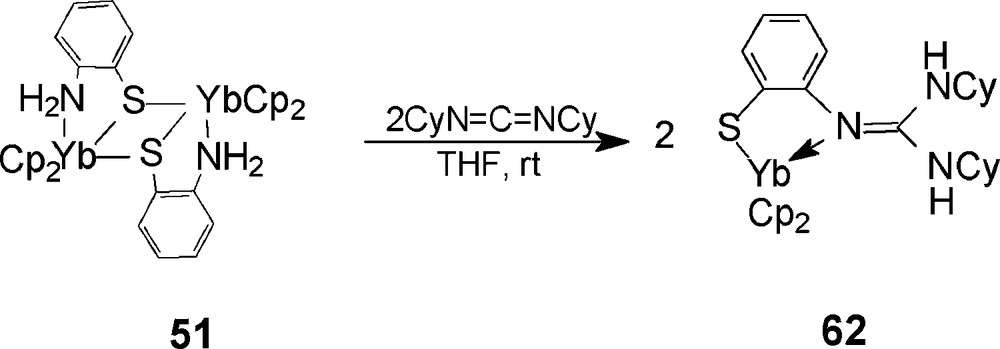
The reaction of [Cp2Yb(o-H2NC6H4S)]2 (51) with CyN=C=NCy has also been studied. Addition of two equivalents of CyN=C=NCy to a THF solution of 51 at ambient temperature results in the formation of the N–H bonds addition product Cp2Yb[SC6H4N=C(NHCy)2] (62) [31], where both hydrogen atoms of the ortho-position amino group are shifted to the nitrogen atoms of the carbodiimide moiety (Scheme 33). X-ray diffraction analysis results indicate that the neutral guanidine substituent is coordinated to the central metal with η1-bonding mode, no elimination of cyclopentadienyl group takes place, which is different from the observation in the reaction of 51 with iPrN=C=NiPr. The reactive discrimination in the two reactions may be attributed to the difference of the nucleophilic ability of the formed neutral guanidine group due to the effect of steric hindrances in the R groups (R = cyclohexyl, isopropyl).
We also synthesized 63 ([Cp2Y(o-H2NC6H4S)]2·2THF) and examined its reactivity toward CyN=C=NCy and iPrN=C=NiPr. Treatment of two equivalents of CyN=C=NCy with 63 in THF at ambient temperature gave a centrosymmetric dimer product [CpY(μ-η2:η1-SC6H4N=C(NHCy)NCy)(THF)]2 (64) (Scheme 34), indicating that an elimination of the cyclopentadienyl group takes place accompanied by the addition of an amino group to the carbodiimide. This is different from the observation in the reaction of 51 and CyN=C=NCy, but similar to that of reaction of 51 with iPrN=C=NiPr. Interestingly, 63 reacts with two equivalents of iPrN=C=NiPr, only CpY(THF)[μ-η2:η1-SC6H4N=C(NHiPr)NiPr)](μ-η2:η1-SC6H4NH2)YCp2·THF (65) is isolated, of which only one amino group adds to the carbodiimide.

When we extend this addition to other lanthanides with larger ion radii, their reactions seem to be more complicated. For example, reaction of CyN=C=NCy with [Cp2Gd(o-H2NC6H4S)]2 (66) in 1:1 ratio gives an interesting co-crystalline compound {Cp2Gd[SC6H4N=C(NHCy)2]}·{CpGd(THF)[μ-η3:η1-SC6H4N=C(NHCy)NCy)][μ-η2:η1-SC6H4NH2]GdCp2·THF} (67), as shown in Scheme 35. No single component through increase of the quantities of carbodiimide or 66 can be isolated in the reactions. The formation of the co-crystalline compound 67 should be the balance of two components of 67a and 67b in the crystallizing process. The results indicate that the NH2 group addition and/or cyclopentadienyl elimination are strongly affected by the factors of the rare earth metal ion character and the steric hindrance of carbodiimides.

To study the sequence of the NH2 addition and Cp elimination, an extension experiment has also been designed. As shown in Scheme 31, the amino group bis-addition product CpYb[SC6H4NC(NHiPr)2]2(THF) (68) readily reacts with one equivalent of Cp3Yb to give a Cp elimination product [CpYb(μ-η2:η1-SC6H4N=C(NHiPr)NiPr)(THF)]2 (55) (Scheme 36). This result indicates that the newly formed guanidine groups can further provide one proton to eliminate the cyclopentadienyl group of Cp3Yb. In other words, the addition of N−H bonds to the C=N bond of carbodiimide molecules must occur prior to the elimination of the cyclopentadienyl group in our observations.

Addition of one equivalent of iPrN=C=NiPr or CyN=C=NCy to the THF solution of Cp2Ln(p-H2NC6H4S)(THF) (52) at room temperature gives the amino group addition products [Cp2Ln(μ-SC6H4N(H)C(NHR)=NR)]4 (R = iPr, Ln = Yb(69a), Er(69b); R = Cy, Ln = Yb(70a), Er(70b)) in moderate yields, indicating that the para-position amino group undergoes an intermolecular addition, to yield the novel square-planar organolanthanide macrocycles, as shown in Scheme 37 [32]. This reaction provides a novel strategy for the construction of square-planar organolanthanide macrocycles by the ligand-based addition of functionalized substituents to unsaturated organic small molecules. It may be attributed to stronger donor electron ability of the formed guanidine group in [p-SC6H4N(H)C(NHR)=NR)] than the free amino group in p-NH2C6H4S. The former is readily coordinated to another lanthanide ion instead of the coordinated THF molecule for construction of the square-planar metallomacrocycles.

Addition of one equivalent of diamine H2N(CH2)3NH2 and iPrN=C=NiPr to the THF solution of Cp3Ln at room temperature gave the novel amino-tethered guanidinate complexes Cp2Ln[H2N(CH2)3N=C(NHiPr)NiPr)] (Ln = Yb(71a), Y(71b), Er(71c), Dy(71d)) in high yields (Scheme 38) [33]. No bis-addition products can be obtained even with the elevated temperature and/or increasing the quantity of carbodiimide. Furthermore, treatment of two equivalents of iPrN=C=NiPr with one equivalent of H2N(CH2)3NH2 and two equivalents of Cp3Yb under the same conditions also gave 71a and residual Cp3Yb, the residual NH2 group cannot further add to the C=N bonds of another carbodiimide molecules [14].

4 Conclusions
It is clear from the above studies that organolanthanide complexes with the N−H bond can serve as a new platform to explore the ligand-based reactions in organolanthanide chemistry via the formal addition of the N−H bond to unsaturated organic molecules. In contrast with the insertion chemistry of the Ln−N bond of organolanthanide amides, these addition reactions are more novel and interesting, and open a new area in organolanthanide chemistry. It should be possible to make further predictions that the development of new reactive and selective organolanthanide complexes with other X−H bonds such as the P−H bonds [34] should lead to more and more novel reaction patterns and organolanthanide derivatives, and the applications of the ligand-based reactions of organolanthanides in organic synthesis will surely increase.
Acknowledgment
We thank the National Natural Science Foundation of China, 973 program (2009CB825300), NSF of Shanghai (09ZR1403300), and Shanghai Leading Academic Discipline Project (B108) for financial support.