

1 Introduction
The self-organisation of discrete units to form supramolecular aggregates and networks has become one of the most active areas of current research [1]. Contrary to established methods in this field which make use of N- and O-donor-containing ligands to connect different metal centres, our strategy has been focused on the use of substituent-free Pn ligand complexes as connecting moieties. This opens new perspectives in the synthesis of phosphorus based inorganic polymers and oligomers [2]. We have broadly used [Cp*Fe(η5-P5)] (Cp* = η5-C5Me5) as a building block to form one-dimensional (1D) and two-dimensional (2D) polymers with Cu(I) [3], Ag(I) [4], and Tl(I) salts [5]. By using special reaction conditions and stoichiometries, the reaction with Cu(I) halides leads to the soluble spherical aggregates with fullerene-like topology [6]. Furthermore, these systems are able to encapsulate smaller guest molecules such as C60 or o-carborane [6a,b]. The ability to form polymeric or oligomeric structures is not limited to [Cp*Fe(η5-P5)]. The Pn ligand complexes [{Cp(CO)2M}2P2(μ,η2-P2)] (M = Cr, Mo) and [(Cp*Mo)2(μ,η3-P3)(μ,η2-PS)] form oligomeric or polymeric aggregates as well [5,7], whereas [Cp″Ta(CO)2(η4-P4)] (Cp″ = η5-C5H3tBu2) forms a fullerene-like spherical cluster revealing a C32 topology [8].
Recently, we introduced the 1,2,4-triphosphaferrocenes [CpRFe(η5-P3C2tBu2)] (CpR = Cp, η5-C5H2tBu3-1,2,4′) and the 1,1′,2,2′,4,4′-hexaphosphaferrocene [Fe(η5-P3C2tBu2)2] as ligands in the coordination chemistry of Cu(I) halides [9]. The hexaphosphaferrocene [Fe(η5-P3C2tBu2)2] reacts with CuX (X = Cl, Br, I) mainly to 1D polymers, e.g. like A containing (CuX)n ladder-like structures or oligomeric complexes with a Cu4Cl4 stair-like core [9b]. In all of these products, hexaphosphaferrocene acts as an additional metal bridging ligand along a (CuX)n polymeric strand. The reaction of the 1,2,4-triphosphaferrocenes [CpRFe(η5-P3C2tBu2)] with CuCl is very intriguing since the nature of the products obtained strongly depends on the steric bulk of the CpR ligand. Thus, the Cp containing complex [CpFe(η5-P3C2tBu2)] reacts with CuCl, independent of the stoichiometry to form the dimeric complex B. The sterically more crowded triphosphaferrocene [Cp‴Fe(η5-P3C2tBu2)] (Cp‴ = η5-C5H2tBu3) forms the dimer C, however, with an excess of CuCl a fragmentation of the P3C2tBu2 ring is observed with formation of the polymeric complex D revealing a novel tetraphosphabutadiene ligand [9a]. Since the bulkiness of the Cp* substituent lies between that of the Cp and Cp‴ ligands, the question arose if the Cp* substituted 1,2,4-triphosphaferrocene [Cp*Fe(η5-P3C2tBu2)] (1) reacts with Cu(I) halides by ligand fragmentation similarly to the Cp‴ derivative or by retaining its structure as in the Cp derivative. Moreover, a search of the so far known coordination modes of the 1,2,4-triphosphaferrocene complex 1 shows that with Fe, Cr, Mo, W [10,11], and Ru carbonyls [12] a mono-coordination mode I was observed, whereas with Ni [11] and Ru carbonyls [12] a dicoordination mode II was found. So far no examples exist in which all three P atoms are incorporated in the coordination behavior represented b mode III.
Herein we report the first examples of type III coordination mode of 1. Moreover, in contrast to the structural relatives of the 1,2,4-triphosphaferrocenes containing a Cp and a Cp‴ ligand, the Cp* derivative 1 tends to react with Cu(I) halides forming oligomeric (CuX)n cages surrounded by moieties of 1.
2 Results and discussion
The slow diffusion reactions of [Cp*Fe(η5-P3C2tBu2)] (1) in toluene with CuX (X = Cl, Br, I) in acetonitrile in a 1:1 molar ratio results in the formation of bright red crystals of the oligomeric compounds [{Cu(μ-X)}6(μ6-X)Cu(MeCN)3{μ,η2-(Cp*Fe(P3C2tBu2))}2{μ3,η3-(Cp*Fe(P3C2tBu2))}] (X = Cl (2), Br (3)), and [{Cu(μ-I)}3{Cu(μ3-I)}3Cu(μ6-I){μ,η2-(Cp*Fe(P3C2tBu2))}3{η1-(Cp*Fe(P3C2tBu2))}] (4) in high yields (Scheme 1). The same products 2–4 are formed when a 1:2 stoichiometry of the reagents is used. Only by using a large excess of CuI (1:4 stoichiometry) is a novel red crystalline 2D polymer [{Cu(μ-I)}4{Cu(μ3-I)(MeCN)}2{μ3,η3-(Cp*Fe(P3C2tBu2))}]n (5) formed (Scheme 1).
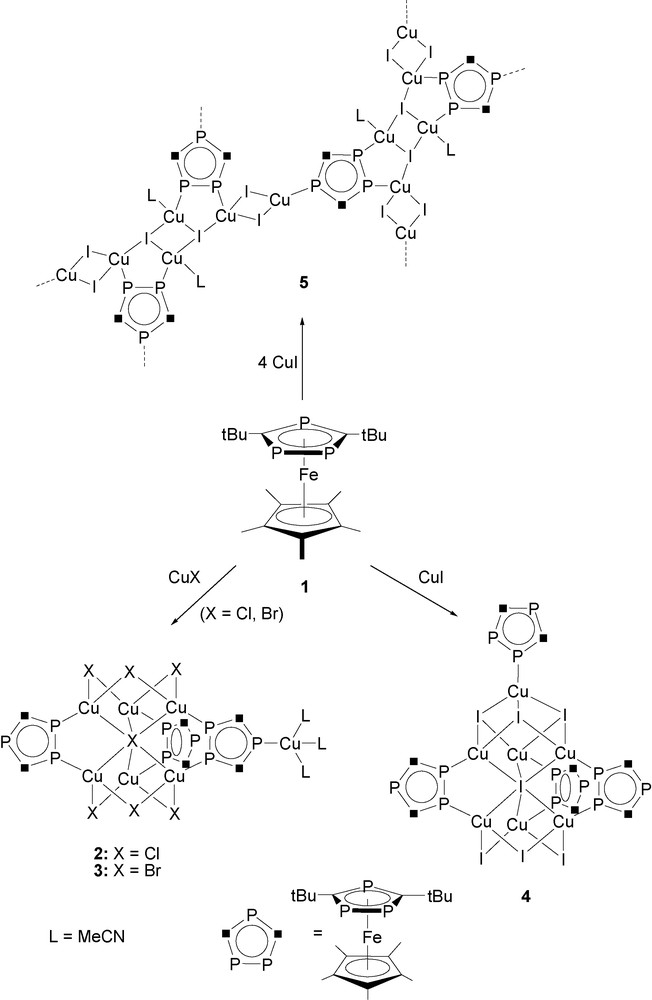
Formation of oligomeric compounds 2, 3, 4, and the 2D polymer (5) by reaction of 1 with CuX (X = Cl, Br, I).
Compounds 2, 3, and 4 are insoluble in nonpolar organic solvents, but 2 and 3 dissolve moderately in a mixture of toluene and acetonitrile. In contrast, 4 dissolve only moderately in polar solvents such as MeCN. In the positive ion mass spectra of 3 obtained by the electro-spray ionization technique (ESI-MS), the peak with the highest mass corresponds to the molecular ion peak diminished by bromide and three MeCN ligands. Moreover, the successive loss of one, two as well as four CuBr fragments is detected. In contrast, in the positive ion ESI-MS spectra of 2 and 4 only fragment peaks are detected revealing [Cp*Fe(η5-P3C2tBu2)}3(CuCl)2Cu]+ and [Cp*Fe(η5-P3C2tBu2)}2(CuI)4Cu(MeCN)]+ units.
Product 5 is insoluble in organic solvents which prevent characterization by solution NMR spectroscopy. In the solid-state 31P{1H} MAS NMR spectrum of 5 three partly overlapping quartets are detected. By simulation the chemical shifts (δ = 37, 18 and 3 ppm) and the 1JCuP coupling constants (1JCuP = 1558 Hz, 1130 Hz and 1286 Hz) are obtained. This indicates that all three phosphorus atoms of 1 are coordinated to a copper atom in 5.
Although the solubility of 2–4 is low it is possible to record the 31P{1H} NMR spectra in CD3CN. In all cases two broad signals in a 1:2 intensity ratio are detected (2: δ = 35.3 ppm (ω1/2 = 116 Hz) and δ = 33.3 ppm (ω1/2 = 440 Hz); 3: δ = 38.3 ppm (ω1/2 = 109 Hz) and 17.9 ppm (ω1/2 = 122 Hz); 4: δ = 45.8 ppm (2JPP = 45.3 Hz) and a broad singlet at δ = 21.2 ppm (ω1/2 = 120 Hz)). In comparison to the starting material, (1) (δ(31P) = 38.1 ppm (d) and 48.2 ppm (t)) [12] these signals are shifted upfield, which indicates the coordination to copper. This is less pronounced for the signal of the unique P atom in 4, which indicates that this P atom is not involved in the dynamic behavior of the molecules in solution, which is evident for the other ones by the observed broad signals. Attempts to investigate this process by variable temperature NMR spectroscopy failed due to the low solubility of the products. The exact nature of this dynamic process is unknown, but for 2 and 3 it seems that all P atoms are incorporated, which probably means that the Cu(MeCN)3 unit migrates via an intra- or intermolecular exchange around the three coordinated triphosphaferrocene units leading to their magnetic equivalency on the NMR time scale at room temperature. For 4, the data indicate that only the adjacent P atoms are involved in the dynamic process in solution.
The molecular structures of 2–4 have been determined by single crystal X-ray diffractions1. Compounds 2 and 3 are isostructural and crystallize isomorphously in the monoclinic space group P21/m. Their molecular structures are depicted in Fig. 1.
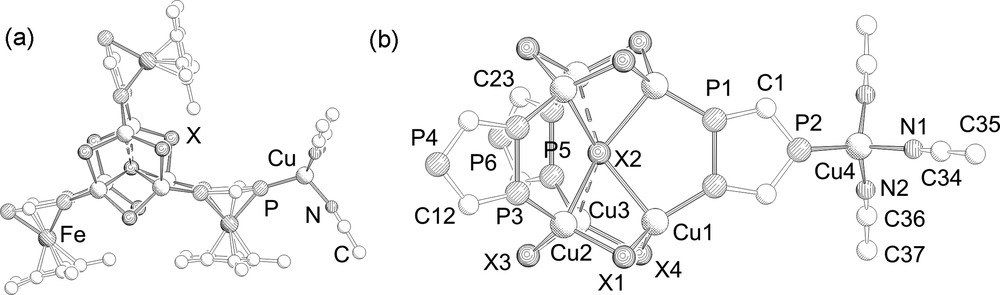
a: Molecular structure of 2 and 3. Hydrogen atoms and tBu-groups are omitted for clarity; b: Central core in 2 and 3. From the 1,2,4-triphosphaferrocene ligands only the P3C2 rings are depicted. Selected bond lengths (Å) and angles (°) for 2 (X = Cl): P1–P1 2.093(2), P3–P3 2.077(2), P5–P5 2.095(2), Cu4–P2 2.260(3), Cu2–P3 2.186(2), Cu3–P5 2.169(2), Cu1–P1 2.188(2), Cu2–Cl3 2.307(2), Cu2–Cl2 2.749(2), Cu3–Cl4 2.244(2), Cu3–Cl2 2.966(1), Cu2–Cl1–Cu1 85.04(6), Cu1–Cl4–Cu3 87.86(7), Cu3–Cl3–Cu2 84.09(7), Cu2–Cl2–Cu1 67.05(4). Selected bond lengths (Å) and angles (°) for 3 (X = Br): P1–P1 2.101(2), P3–P3 2.094(2), P5–P5 2.102(2), Cu4–P2 2.262(3), Cu2–P3 2.212(3), Cu3–P5 2.187(2), Cu1–P1 2.213(2), Cu2–Br3 2.430(2), Cu2–Br2 2.844(2), Cu3–Br4 2.373(2), Cu3–Br2 2.939(1), Cu2–Br1–Cu1 81.66(5), Cu1–Br4–Cu3 83.34(5), Cu3–Br3–Cu2 80.76(5), Cu2–Br2–Cu1 66.95(4).
The solid-state structure of 2 and 3 has two (CuX)3 six-membered rings which are connected through a μ6 coordinating X ligand and are additionally bridged by three 1,2,4-triphosphaferrocene units via the adjacent phosphorus atoms. The counter ion is coordinated by the unique phosphorus atom of the 1,2,4-triphosphaferrocene in a Cu(MeCN)3 fragment. This triphospholyl moiety represents the novel coordination mode III. The copper atoms show distorted tetrahedral coordination geometry. Interestingly, the coordination of the μ6-X ligand to the six copper atoms is not symmetrical. Four shorter Cu–(μ6-X) (2: Cu–Cl 2.749(2)–2.790(2) Å; 3: Cu–Br 2.844(1)–2.838(1) Å) and two longer distances (2: Cu–Cl 2.966(2); 3: Cu–Br 2.939(1) Å) are formed. Although the longer Cu–X distances are relatively long compared to usual Cu–X distances found, they are shorter than the sum of the van der Waals radii of the corresponding elements (ΣvdW (CuCl) = 3.15 Å, ΣvdW (Cu–Br) = 3.25 Å) [13]. It is likely that the distance between the two (CuX)3 rings was determined by the chelating triphosphaferrocene ligands causing the longer Cu–X distances. In the Cu(II) pyrazolate complex [Cu6(μ6-Cl)(MeO)2(pz)9] (pz = pyrazole) [14], which also contains a μ6-Cl ligand, the Cu–Cl distances (2.623(2) and 2.603(1) Å) are shorter than those found in 2. The Cu–(μ2-X) distances in 2 and 3 vary slightly (2: Cu–Cl 2.243(2)–2.307(2) Å; 3: Cu–Br 2.372(1)–2.435(1) Å) and as one would expect they are considerably shorter than the corresponding Cu–(μ6-X) distances. The Cu–P distances of the bridging triphosphaferrocene ligands are slightly shorter for 2 (2.169(2)–2.188(2) Å) in comparison to 3 (2.188(1)–2.213(2) Å) whereas the Cu–P distance of the unique phosphorus atom is equal and within the experimental error (2: 2.260(3) Å; 3: 2.262(3) Å).
41.4 C7H8MeCN crystallizes as red cubic crystals in the triclinic space group . The molecular structure is depicted in Fig. 2.
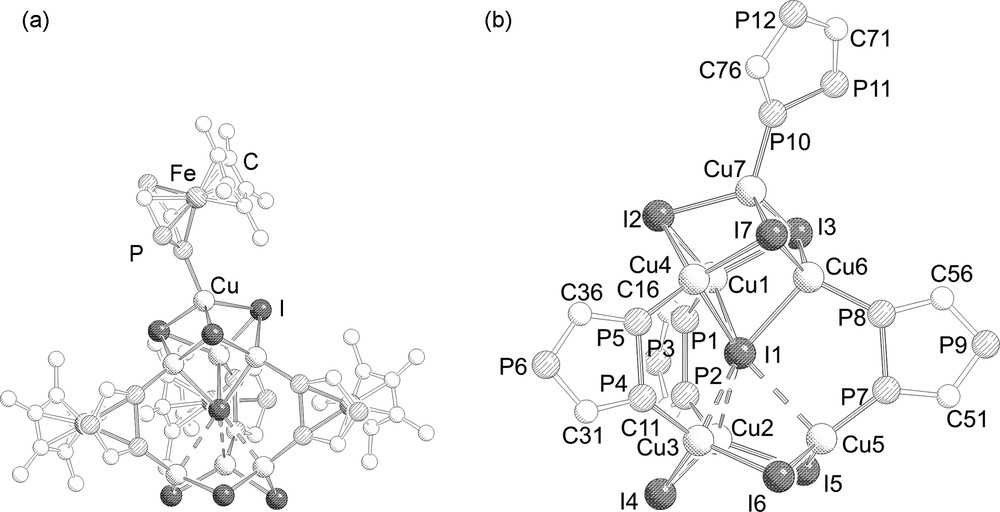
a: Molecular structure of 4. Hydrogen atoms and tBu groups are omitted for clarity; b: central core in 4. Cp*Fe moieties are omitted additionally. Selected bond lengths (Å) and angles (°): Cu1–I2 2.6256(5), Cu6–I1 2.8291(5), Cu6–I7 2.6015(5), Cu4–I7 2.6640(4), Cu3–I4 2.5714(5), Cu5–I5 2.5500(5), Cu7–P10, 2.2284(9), Cu6–P8 2.239(1), Cu5–P7 2.2258(9), Cu3–P4 2.232(1), P10–P11 2.106(1), P7–P8 2.096(1), P1–P2 2.091(1), P4–P5 2.099(1), Cu1–P1–P2 118.94 (4), Cu1–I1–Cu6 72.42(1), Cu7–I3–Cu6 66.14(1), I6–Cu5–I5 120.05(2), I1–Cu4–I2 109.93(1).
The molecular structure of 4 is similar to those of 2 and 3 with the main difference being that the Cu counter ion for the interstitial iodide atom is incorporated in a Cu4I4 cube and additionally end-on coordinated by a [{Cp*Fe(η5-P3C2tBu2)}] moiety instead of binding to a triphosphaferrocene as observed in 2 and 3. Alternatively, the structure of 4 is described as consisting of two (CuI)4 cubes sharing a joint corner, whereas from one of the (CuI)4 cubes one corner (opposite to the shared corner) is missing. This central core is then stabilized by the coordination of three triphospholyl ligands through the adjacent phosphorus atoms (type II) bridging the (CuI)4 cubic unit and the (CuI)3 six-membered ring. The apical copper atom is additionally coordinated by a triphosphaferrocene ligand in a η1 fashion through one of the two adjacent phosphorus atoms (type I). Similarly, as in 2 and 3, the Cu–(μ6-I) distances are divided in three shorter (2.742(2)–2.829(1) Å) and three longer ones (2.998(2)–3.258(1) Å), but even the longer distances are shorter than the van der Waals radii of copper and iodine (ΣvdW = 3.38 Å) [13]. As one would expect the Cu–I distances decrease by going from μ3-I (2.601(1)–2.750(2) Å) to μ2-I (2.536(2)–2.561(1) Å). The Cu–P bond length involving the bridging triphospholyl units differ slightly (2.217(2)–2.244(1) Å) and are similar to the Cu–P bond length (2.228(2) Å) of the η1 coordinated triphosphospholyl moiety.
By analyzing the distances between the copper atoms which are bridged by the triphospholyl units in the complexes 2–4 (2: 4.288(1)–4.618(2) Å, 3: 4.413(1)–4.539(1) Å and 4: 4.429(3)–4.452(3) Å) an alternative description of these complexes emerge. The two (CuX)3 six-membered rings in 2 and 3 are held together by the triphosphaferrocene ligands leading to the formation of a cavity in which the corresponding anion is accommodated. Since the size of the cavity is determined by the triphosphaferrocene moieties, and obviously it is larger than the anions would require, relatively long Cu–X distances are formed. The Cu counter ion is either attached to the unique phosphorus atom of the triphospholyl ligands or, as in 4, incorporated into the (CuI)4 cage.
Complex 5 crystallizes as ruby plates in the monoclinic space group P21/c. The molecular structure is depicted in Fig. 3.
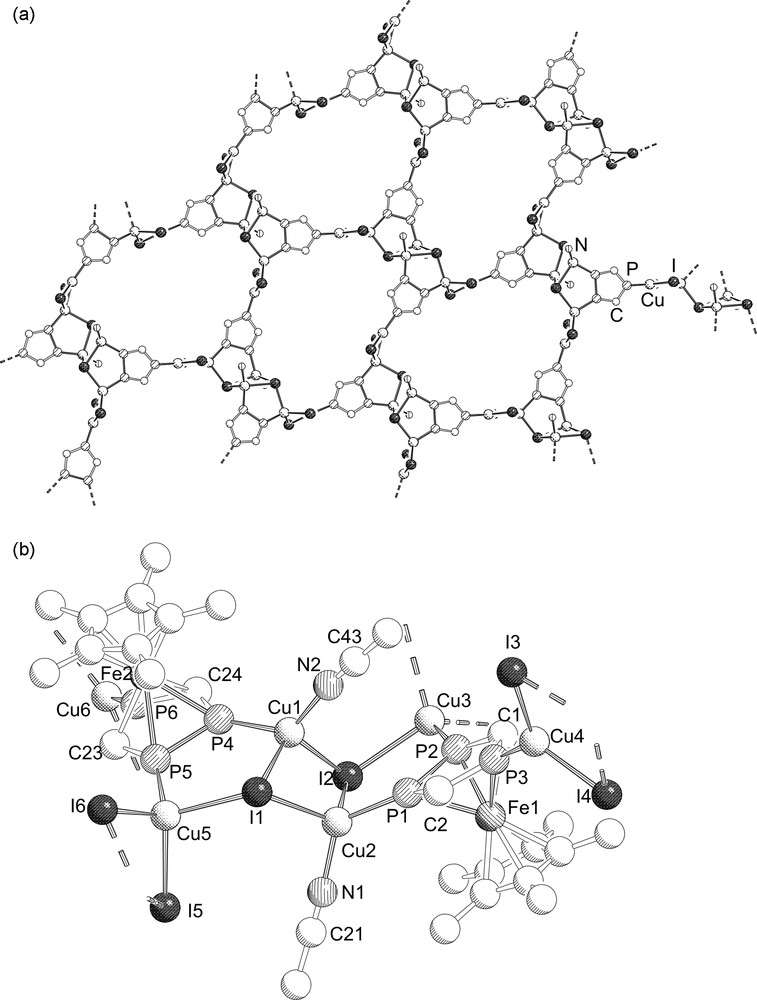
a: Section of the 2D polymeric structure of 5. Hydrogen and Cp*Fe units are omitted for clarity; b: Repeating unit of the polymeric structure of 5. The hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): I(1)–Cu(1) 2.6980(17), I(1)–Cu(2) 2.6638(18), I(1)–Cu(5) 2.671(2), I(2)–Cu(1) 2.6416(17), I(2)–Cu(2) 2.7107(17), I(2)–Cu(3) 2.669(2), I(3)–Cu(4) 2.5138(18), I(3)–Cu(3)#1 2.6127(19), I(4)–Cu(4) 2.5097(19), I(4)–Cu(3)#1 2.6763(18), I(5)–Cu(5) 2.6477(19), I(5)–Cu(6)#2 2.5058(17), I(6)–Cu(5) 2.6475(19), I(6)–Cu(6)#2 2.5549(18), Cu(1)–P(4) 2.215(4), Cu(1)–N(2) 1.974(12), Cu(2)–P(1) 2.240(4), Cu(2)–N(1) 1.959(13), Cu(3)–P(2) 2.248(3), Cu(4)–P(3) 2.198(4), Cu(5)–P(5) 2.248(3), Cu(6)–P(6) 2.197(3), P(1)–P(2) 2.094(5), P(1)–C(2) 1.763(12), P(2)–C(1) 1.781(13), P(3)–C(1) 1.747(12), P(3)–C(2) 1.718(14), P(4)–P(5) 2.091(5), P(4)–C(24) 1.756(11), P(5)–C(23) 1.743(12), P(6)–C(23) 1.754(12), P(6)–C(24) 1.740(14); I(1)–Cu(1)–I(2) 114.85(6), I(1)–Cu(1)–P(4) 100.96(11), I(2)–Cu(1)–P(4) 103.85(9), I(1)–Cu(2)–I(2) 113.69(6), I(1)–Cu(2)–P(1) 111.83(10), I(2)–Cu(2)–P(1) 96.44(10), I(2)–Cu(3)–P(2) 97.42(11), I(2)–Cu(3)–I(3)#3 114.56(6), I(2)–Cu(3)–I(4)#3 109.90(7), I(3)#3–Cu(3)–P(2) 120.20(12), I(4)#3–Cu(3)–P(2) 107.95(10), I(3)#3–Cu(3)–I(4)#3 106.40(6), I(3)–Cu(4)–I(4) 114.94(7), I(3)–Cu(4)–P(3) 125.08(11), I(4)–Cu(4)–P(3) 119.93(11), I(1)–Cu(5)–I(5) 110.10(6), I(1)–Cu(5)–I(6) 108.93(6), I(1)–Cu(5)–P(5) 101.00(12), I(5)–Cu(5)–I(6) 109.22(7), I(5)–Cu(5)–P(5) 110.91(10), I(6)–Cu(5)–P(5) 116.34(11), I(5)#4–Cu(6)–P(6) 133.85(11), I(6)#4–Cu(6)–P(6) 108.97(10), I(5)#4–Cu(6)–I(6)#4 117.07(6), Cu(2)–P(1)–P(2) 120.13(16), Cu(3)–P(2)–Fe(1) 145.53(16), Cu(3)–P(2)–P(1) 118.3(2), Cu(3)–P(2)–C(1) 138.9(4), P(1)–P(2)–C(1) 100.4(4), Cu(4)–P(3)–Fe(1) 142.68(16), Cu(4)–P(3)–C(1) 128.8(5), Cu(4)–P(3)–C(2) 126.2(4), C(1)–P(3)–C(2) 103.7(6), Cu(1)–P(4)–Fe(2) 140.92(15), Cu(1)–P(4)–P(5) 118.03(16), Cu(1)–P(4)–C(24) 140.7(5), Cu(5)–P(5)–Fe(2) 143.70(16), Cu(5)–P(5)–P(4) 116.79(18), Cu(5)–P(5)–C(23) 140.7(5), Cu(6)–P(6)–C(23) 127.6(5), Cu(6)–P(6)–C(24) 125.4(4). Symmetry transformations used to generate equivalent atoms: #1 = x + 1, y − 1, z; #2 = x, y − 1, z; #3 = x + 1, y, z; #4 = x, y, z.
The structure of 5 is determined by single crystal X-ray diffraction. The solid-state structure is depicted in Fig. 3. The central core of 5 consists of a (CuI)6 chain built up from three (CuI)2 four-membered rings which are bonded to each other by Cu–I bonds. Two triphospholyl ligands bridge two consecutive (CuI)2 four-membered rings by coordination to copper atoms via the adjacent phosphorus atoms (type III). The unique phosphorus atom coordinates to a neighboring (CuI)6 chain leading to a two dimensional architecture (Fig. 3a). Each triphosphaferrocene moiety is coordinated to three different copper atoms. The copper atoms connected to the unique phosphorus atoms are trigonal planar whereas the other copper atoms are tetrahedrally coordinated. The fourth coordination site in the central (CuI)2 unit of the (CuI)6 chain is occupied by MeCN ligands. The Cu–I distances for the trigonal-planar coordinated copper atoms are slightly shorter (2.511(1)–2.516(1) Å) than the corresponding distances for the tetrahedrally coordinated copper (2.646(1)–2.715(1) Å). A similar trend is observed for the Cu–P distances (Cu–Padjacent = 2.219(1)–2.248(1) Å and Cu–Punique = 2.204(3)–2.205(1) Å), although it is not so pronounced as for the Cu–I distances.
3 Conclusions
We have shown that the Cp* containing 1,2,4-triphosphaferrocene [Cp*Fe(η5-P3C2tBu2)] reacts with CuX (X = Cl, Br, I) without fragmentation to act as chelating ligands surrounding (CuX)n cages. The oligomeric complexes 2–4 are obtained by using 1:1 or 1:2 stoichiometries whereas the 2D polymer 5 is formed with an excess of CuI. The cages containing products 2–4 show two (CuX)3 six-membered rings connected by bridging triphospholyl ligands of the 1,2,4-triphosphaferrocenes. The counter ion is either coordinated additionally as a Cu(CH3CN)3 unit at the unique P atom of the triphospholyl ring (2 and 3) or by an incorporated Cu atom into the formed (CuI)3 moiety as in the case of the iodide derivative 4. The cage stabilization properties of the Cp* containing triphosphaferrocene 1 are different from the, so far observed, features of the Cp and Cp”’ analogues of 1. Moreover, a novel 1,2,4-coordination mode (type III) of the 1,2,4-triphosphaferrocene is found in this oligomeric derivative besides the previously found 1,2- and 1-coordination modes. Moreover, by increasing stoichiometry of the reactants to the large excess of CuI, a novel 2D polymer 5 is formed instead of oligomer 4. This polymer, exclusively, reveals the 1,2,4-coordination mode (type III) of the bridging triphospholyl ligands of the 1,2,4-triphosphaferrocenes.
4 Experimental section
All manipulations are carried out under an atmosphere of dinitrogen using standard Schlenk techniques. All solvents are dried using standard procedures and freshly distilled before use. [Cp*Fe(η5-P3C2tBu2)] is prepared according to the literature [12]. Solution NMR spectra were recorded at 25 °C on a Bruker Avance 400 (1H: 400.132 MHz, standard tetramethylsilane, 31P: 161.976 MHz, standard 85% H3PO4). MAS NMR spectra were recorded on a Bruker Avance 300 spectrometer [1H: 300.123 MHz, 31P: 121.495 MHz standard NaH2PO4 (2.3 ppm)] by using a double resonance 2.5 mm MAS probe. The spectra were acquired at a MAS rotation frequency of 30 kHz with a 90° pulse length of about 2.5 μs and a relaxation delay of 300 s (31P) and 10 s (1H).
Synthesis of [{Cu(μ-Cl)}6(μ6-Cl)Cu(MeCN)3{μ,η2-(Cp*Fe(P3C2tBu2))}2{μ3,η3-(Cp*Fe(P3C2tBu2))}] (2): A solution of CuCl (16 mg, 0.158 mmol) in 10 mL of CH3CN was layered onto a solution of [Cp*Fe(η5-P3C2tBu2)] (2) (67 mg, 0.158 mmol) in 10 mL of toluene at room temperature. After complete diffusion of phases, the reaction mixture is filtered, reduced to one half of its amount and placed in an undisturbed area at room temperature. Bright red plates of 2 are formed. The mother liquor is decanted and the obtained crystals are washed with pentane and dried under vacuum. Yield: 33 mg (0.015 mmol, 64%).
31P{1H} NMR (C6D6/toluene/CH3CN): δ = 35.3 ppm (broad, ω1/2 = 116 Hz), 33.3 (broad, ω = 440 Hz). ESI-MS (positive ion) m/z (%): 1527 (70) [{Cp*Fe(η5-P3C2tBu2)}3(CuCl)2Cu]+, 1429 (100) [{Cp*Fe(η5-P3C2tBu2)}3(CuCl)Cu]+, 1007 (27) [{Cp*Fe(η5-P3C2tBu2)}2(CuCl)Cu]+, 907 (67) [{Cp*Fe(η5-P3C2tBu2)}2Cu]+, 526 (74) [{Cp*Fe(η5-P3C2tBu2)}2Cu(MeCN)]+. ESI-MS (negative ion) m/z (%): 135 (100) [CuCl2]−. Elemental analysis calculated for C66H108Cl7Cu7P9Fe3N3 (2082.8 g/mol) C 38.055%, H 5.23%, N 2.02. Found C 38.12%, H 5.17%, N 1.69%.
Synthesis of [{Cu(μ-Br)}6(μ6-Br)Cu(MeCN)3{μ,η2-(Cp*Fe(P3C2tBu2))}2{μ3,η3-(Cp*Fe(P3C2tBu2))}] (3): A solution of CuBr (46 mg, 0.32 mmol) in 10 mL of CH3CN is layered onto a solution of [Cp*Fe(η5-P3C2tBu2)] (1) (67 mg, 0.158 mmol) in 10 mL of toluene at room temperature. After complete diffusion of phases, the reaction mixture is filtered, reduced to one half of its amount and placed in an undisturbed area at room temperature. After a few days bright red plates of 3 are formed. The mother liquor is decanted and the obtained crystals are washed with pentane and dried under vacuum. Yield: 63 mg (0.026 mmol, 57.5%).
1H NMR (CD2Cl2): δ = 1.41 (s, 15 H; C5(CH3)5), 1.94 (s, 18 H, C(CH3)3), 1.98 (s, 9 H, CH3CN). 31P{1H} NMR (CD2Cl2): δ = 38.3 ppm (broad, ω1/2 = 109 Hz, 1P), 17.9 (broad, ω = 122 Hz), 2P). ESI-MS (positive ion) m/z (%): 2191 (12) [{Cp*Fe(η5-P3C2tBu2)}3(CuBr)6Cu]+, 2047 (26) [{Cp*Fe(η5-P3C2tBu2)}3(CuBr)5Cu]+, 1905 (11) [{Cp*Fe(η5-P3C2tBu2)}3(CuBr)4Cu]+, 1339 (20) [{Cp*Fe(η5-P3C2tBu2)}2(CuBr)3Cu]+, 1195 (48) [{Cp*Fe(η5-P3C2tBu2)}2(CuBr)2Cu]+, 670 (100) [{Cp*Fe(η5-P3C2tBu2)}(CuBr)Cu(MeCN)]+, 526 (70) [{Cp*Fe(η5-P3C2tBu2)}Cu(MeCN)]+. ESI-MS (negative ion) m/z (%): 940 (4) [Cu6Br7]−, 798 (6) [Cu5Br6]−, 654 (6) [Cu4Br5]−, 511 (27) [Cu3Br4]−, 367 (72) [Cu2Br3]−, 223 (100) [CuBr2]−. Elemental analysis calculated for C66H108Br7Cu7P9Fe3N3 (2267 g/mol) C 33.13%, H 4.55%, N 1.76%. Found: C 33.31%, H 4.66%, N 1.48%.
Synthesis of [{Cu(μ-I)}3{Cu(μ3-I)}3Cu(μ6-I){μ,η2-(Cp*Fe(P3C2tBu2))}3{η1-(Cp*Fe(P3C2tBu2))}] (4): A solution of CuI (31 mg, 0.166 mmol) in 10 mL of CH3CN is layered onto a solution of [Cp*Fe(η5-P3C2tBu2)] (1) (70 mg, 0.166 mmol) in 10 mL of toluene at room temperature. After complete diffusion of the phases, the reaction mixture is filtered, reduced to half of its volume and placed in an undisturbed area at room temperature. After a few days red cubic crystals of 4 are formed. The mother liquor is decanted and the obtained crystals are washed with pentane and dried under vacuum. Yield: 54 mg (0.017 mmol, 71.7%).
1H NMR (CD3CN): δ = 1.40 ppm (s, 9 H; C(CH3)3), 1.93 (s, 15 H; C5(CH3)5). 31P{1H} NMR (CD3CN): δ = 21.2 ppm (broad ω1/2 = 120 Hz, 2 P), 45.8 (t, 2JPP = 45.3 Hz, 1 P). ESI-MS (positive ion) m/z (%): 1712 (2) [{Cp*Fe(η5-P3C2tBu2)}2(CuI)4Cu(MeCN)]+, 1289 (3) [{Cp*Fe(η5-P3C2tBu2)}2(CuI)2Cu]+, 1097 (3) [{Cp*Fe(η5-P3C2tBu2)}2(CuI)Cu]+, 907 (7) [{Cp*Fe(η5-P3C2tBu2)}2Cu]+, 580 (50) [Cp*Fe(η5-P3C2tBu2)Cu(C7H8)]+, 526 (28) [Cp*Fe(η5-P3C2tBu2)Cu(MeCN)]+, 484 (100) [Cp*Fe(η5-P3C2tBu2)Cu]+. ESI-MS (negative ion) m/z (%): 699 (3) [Cu3I4]−, 507 (12) [Cu2I3]−, 317 (100) [CuI2]−, 127 (13) [I]−. Elemental analysis calculated for C90.5H144Cu7I7Fe4P12 (3160.4 g/mol): C 34.39%, H 4.59%. Found: C 35.17%, H 4.89%.
Synthesis of [{Cu(μ-I)}4{Cu(μ3-I)(MeCN)}2{μ3,η3-(Cp*Fe(P3C2tBu2))}]n (5): A solution of CuI (72 mg, 0.38 mmol) in 10 mL of CH3CN is layered onto a solution of [Cp*Fe(η5-P3C2tBu2)] (1) (40 mg, 0.095 mmol) in 10 mL of toluene at room temperature. After complete diffusion of phases, the red colored clear reaction mixture is filtered, reduced to two third of its volume and left in an undisturbed area at room temperature. After two weeks red crystals of 5 are formed. The mother liquor is decanted and the obtained crystals dried under vacuum. Yield: 43 mg (0.022 mmol, 44%). 1H MAS NMR: δ = 1.7 ppm (broad). 31P MAS NMR: δ = 37.38 ppm (q, 1JPCu = 1558 Hz, 1P), 18.02 (q, 1JPCu = 1130 Hz, 1P), 3.43 (q, 1JPCu = 1286 Hz, 1P). 13C MAS NMR: δ = 15.7 (s, C5(CH3)5), 91.4 (s, C5(CH3)5), 37.1 (s, C(CH3)3), 39.5 ppm (s, C(CH3)3). Elemental analysis calculated for C176Cu24Fe8I24N8P24 (1996.16 g/mol): C 25.3%, H 3.67%, N 1.34%. Found: C 25.52%, H 3.37%, N 1.26%.
Acknowledgements
The authors thank the Deutsche Forschungsgemeinschaft (DFG) for financial support. The COST action CM0802 PhoSciNet has supported this work. Prof. J.F. Nixon is gratefully acknowledged for helpful discussions.
1 The crystal structure measurements were carried out on an Oxford Diffraction Gemini R Ultra CCD diffractometer with CuKα-radiation (λ = 1.54180 Å) for 2–4 and on a Stoe IPDS diffractometer with MoKα-radiation (λ = 0.71073 Å) for 5. Crystallographic Data for: 22C7H8, empirical formula C66H108Cl7Cu7Fe3N3P9, Mw. 2267.04, crystal size [mm] 0.38 × 0.12 × 0.05, T = 123 K, λ = 1.54178 Å, monoclinic, P 21/m, a = 16.0573(3) Å, b = 19.2171(3) Å, c = 16.4713(3) Å, β = 101.180(2)°, V = 4986.17(16) Å3, Z = 2, μ = 8.37 mm−1, θ range = 2.73–62.17°, reflections collected/unique 20676/7881, observed reflections with [I > 2σ(I)] 6323, Rint 0.042, final R indices [I > 2σ(I)] R1 = 0.0648, wR2 = 0.1655, R indices (all data) R1 = 0.0786, wR2 = 0.1776. 3, empirical formula C66H108Br7Cu7Fe3N3P9, Mw. 2393.99, crystal size [mm] 0.24 × 0.08 × 0.06, T = 123 K, λ = 1.54178 Å, monoclinic, P 21/m, a = 16.1571(5) Å, b = 19.6483(3) Å, c = 16.4173(4) Å, β = 102.333(3)°, V = 5091.6(2) Å3, Z = 2, μ = 9.676 mm−1, θ range = 2.76–66.50°, reflections collected/unique 14625/7317, observed reflections with [I > 2σ(I)] 5159, Rint 0.0311 final R indices [I > 2σ(I)] R1 = 0.0476, wR2 = 0.1278, R indices (all data) R1 = 0.0669, wR2 = 0.1417. 41.5 C7H8MeCN, empirical formula C80H132Cu7Fe4I7P12, Mw 3201.23, crystal size [mm] 0.16 × 0.16 × 0.12, T = 150 K, λ = 0.71073 Å, monoclinic, , a = 15.9109(3) Å, b = 16.1452(2) Å, c = 24.2355(4) Å, α = 94.459(1)°, β = 93.949(1)°, γ = 109.906(1)°, V = 5805.78(17) Å3, Z = 2, μ = 3.804 mm−1, θ range = 3.0–29.1°, reflections collected/unique 74896/25606, observed reflections with [I > 2σ(I)] 19157, Rint 0.033, final R indices [I > 2σ(I)] R1 = 0.0296, wR2 = 0.0546, R indices (all data) R1 = 0.0524, wR2 = 0.0585. 5, empirical formula C44H72Cu6Fe2I6N2P6, Mw. 2069.26, crystal size [mm] 0.16 × 0.12 × 0.05, T = 123 K, λ = 0.71073 Å, monoclinic, P21/c, a = 20.446(3) Å, b = 18.1755(14) Å, c = 19.138(3) Å, β = 116.890(13)°, V = 6343.0(16) Å3, Z = 4, μ = 5.517 mm−1, θ range = 2.13–25.96°, reflections collected/unique 76757/12290, observed reflections with [I > 2σ(I)] 5588, Rint 0.1168, final R indices [I > 2σ(I)] R1 = 0.0513, wR2 = 0.1184, R indices (all data) R1 = 0.1143, wR2 = 0.1384. CCDC-772698-772701 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request.cif.