1 Introduction
Phosphonium salts are readily available at low cost, stable to oxygen and to moisture, and therefore can be stored under air atmosphere for long periods of time without any detectable deterioration. Their excellent thermal and chemical stability and their non-toxicity make phosphonium derivatives very attractive, and therefore the scope of their applications is large [1]. They have been extensively used as intermediates in organic syntheses as in Wittig reactions [2]. Phosphonium cation-based ionic liquids (ILs) offer in some chemical transformations superior properties than the nitrogen cation-based ILs [3]. The range of applications of these interesting materials, recently investigated, includes their use as extraction solvents, chemical synthesis solvents, electrolytes in batteries and super-capacitors, and in corrosion protection [4]. The most prominent catalytic application of these compounds is phase-transfer catalysis [5]. It has also been demonstrated that phosphonium salts can be used interchangeably with the corresponding phosphines in a broad spectrum of processes ranging from catalytic applications (palladium-catalyzed couplings, acylations of alcohols, and Baylis-Hillman reactions) to stoichiometric transformations (reductions of disulfides and azides) [6]. Phosphonium salts are good activating agents for performing cross-coupling palladium catalyzed arylation [7]. This class of phosphorus compounds was evaluated as powerful catalysts in the Halex reaction [8] which is, to date, one of the best and least expensive ways to introduce fluorine into a molecule. A number of successful applications of phosphonium salts as organocatalysts have been described recently [9] but the Lewis acidic nature of the phosphonium salt catalysts in organic reactions has not yet been fully evaluated.
We have previously reported the synthesis of N−phosphino formamidines of general structure iPr2N−C(H)=N−PR2 [10]. Considering the increasing interest devoted to phosphonium compounds, we decided to broaden our research field to the development of a new class of functionalized phosphonium compounds, the N-phosphonio formamidines of the general formula [iPr2N−C(H)=N−P(R’)R2]+X−. These compounds have been structurally characterized by X-ray diffraction analyses. Their behavior in the presence of a large variety of organic and inorganic bases has been investigated.
2 Results and discussion
N-phosphonio formamidines 2a,b were prepared in good yields in a one-pot procedure after treatment of the corresponding N−phosphino formamidines 1a,b derivatives with MeI in CH2Cl2 at –78 °C (Scheme 1). After 18 hours at reflux in neat 2-bromopropane, the N-phosphonio formamidine 3b was obtained from the corresponding formamidine precursor 1b in quantitative yield based on 31P NMR spectroscopy. The phosphorus-arylation reaction conducted in the presence of Pd(OAc)2 as catalyst on formamidines 1a,b affording the N-phosphonio formamidines 4a,b was adapted from a procedure described by Migita and al. [11]1. Spectroscopic features allow for complete identification of N-phosphonio formamidines 2a,b, 3b and 4a,b.
Mass spectrometry analyses allowed us to identify the molecular peak [M–X]+ (X = I or Br) for all the compounds 2a,b, 3b and 4a,b. FT-IR spectra of these compounds displayed an absorption band between 1597 and 1616 cm−1 corresponding to the ν(C=N) stretching of the formamidine pattern. The 31P NMR chemical shifts for N-phosphonio formamidines 2a,b, 3b and 4a,b are consistent with the characteristic shifts measured for tetracoordinated phosphorus σ4-P fragments (δ ≈ 30–32 ppm [R = Ph], δ ≈ 52–56 ppm [R = iPr]) [10]. The 1H and 13C NMR spectra of the N-phosphonio formamidines showed the presence of the proton and the carbon of the formamidine framework >N−C(H)=N− in the range of 7.9–8.8 ppm and 158–160 ppm and confirmed the presence of the different alkyl and aryl groups attached to the phosphorus atom. Moreover, the low magnitude of the 2JCP coupling constant observed between the imino carbon atom and the phosphorus fragment (3.4 < 2JCP < 7.7 Hz) is characteristic for tetracoordinated σ4-P N-phosphorus formamidino derivatives [12]. 2D HMBC 1H-15N and HMQC 31P-15N{1H} NMR experiments monitored on 2b, 3b and 4a,b allowed us to identify the chemical shift of the imino nitrogen atom of the formamidine fragment (δ 15N – 235–250.0 ppm with a 2JNP ranging from 28 to 38 Hz) which correlates with the corresponding phosphorus atom. It is interesting to note that the chemical shifts of the imino and amino nitrogen atoms in the formamidine pattern are closer to each other in the N-phosphonio formamidines 2b, 3b and 4a,b (Δδ 15N < 40 ppm) than the ones recorded for N−phosphino formamidines 1a,b (Δδ 15N > 60 ppm). This reflects a more pronounced delocalisation of the π-electrons in the N-phosphonium formamidine derivatives (Table 1).
NMR Spectroscopic data for N-phosphonio formamidines 1a,b, 2a,b, 3b and 4a,b.
| Products | δ 31P | δ 1HCH=N (3JHP) | δ 13CC=N (2JCP) | δ 15N (1JNP; N–P) | δ 15N iPr2N |
| 1a | 54.3 | 8.14 (18.9) | 158.6 (52.7) | –182.2 (44.6) | –243.9 |
| 1b | 90.0 | 7.88 (17.4) | 158.2 (47.9) | –177.8 (39.4) | –240.1 |
| 2a | 32.2 | 8.25 (21.8) | 159.3 (7.7) | \ | \ |
| 2b | 56.6 | 8.41 (19.7) | 159.7 (3.4) | –239.6 (37.1) | –216.9 |
| 3b | 56.0 | 8.74 (17.1) | 160.3 (4.5) | –245.8 (37.7) | –215.6 |
| 4a | 30.3 | 7.88 (21.0) | 157.8 (6.8) | –235.4 (27.8) | –211.9 |
| 4b | 51.9 | 8.75 (17.4) | 160.1 (6.4) | –250.5 (32.5) | –213.4 |
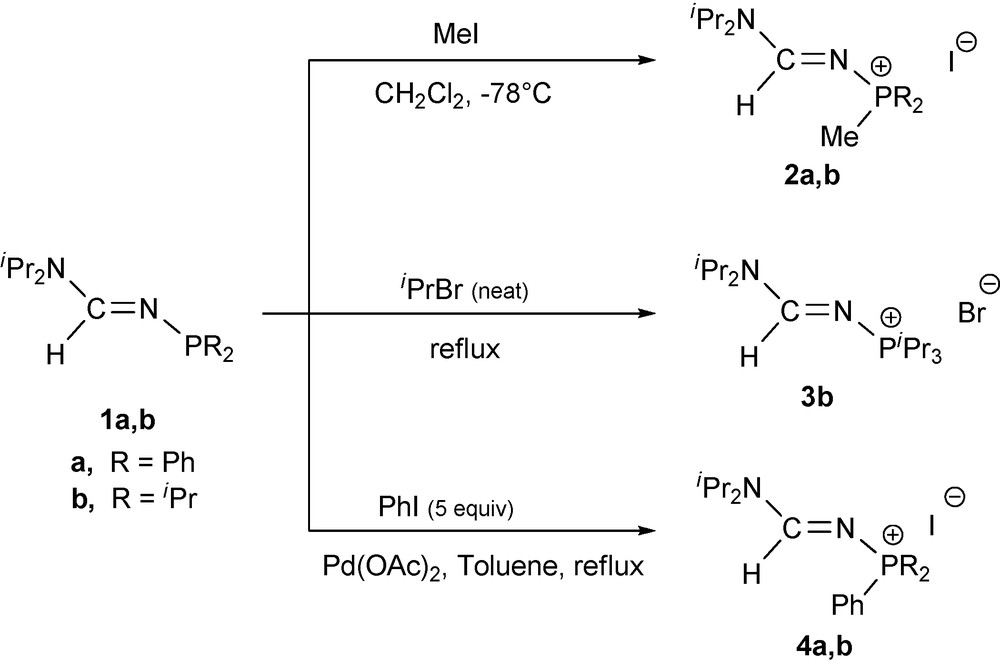
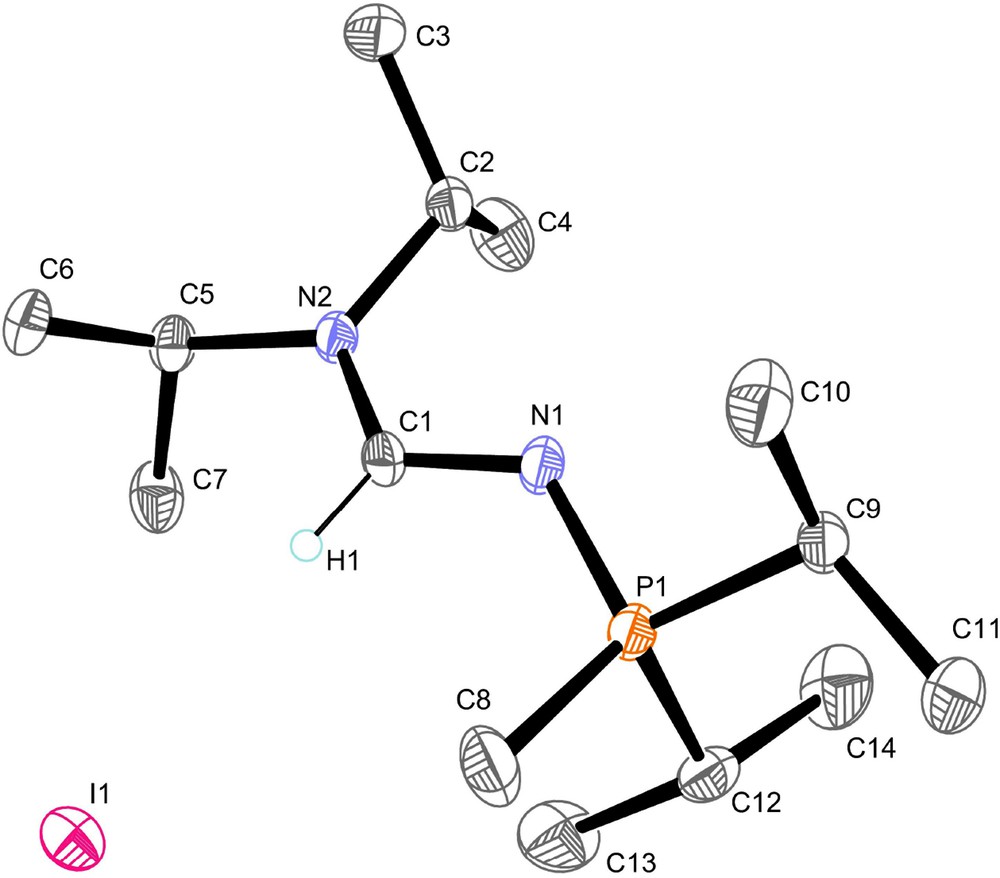
Suitable crystals of compounds 2b and 4a were grown and the single-crystal X-ray diffraction studies revealed an E-formamidine arrangement for both compounds (Figs. 1 and 2). The compounds exhibit very similar structural geometry with a pyramidal phosphorus atom and a planar amino nitrogen atom iPr2N−. The C1−N1 and N1−P1 bond distances of 1.31–1.33 and 1.60–1.61 Å respectively, fall in the range between carbon−nitrogen [13] and nitrogen-phosphorus double and single bonds. There is not a significant difference between the C1−N1 and C1−N2 bond lengths, which denotes a strong electronic delocalisation along the formamidine NC(H)N moiety. In comparison with the structure of the N−phosphino formamidine 1a, the N2−C1−N1 angle value in 2b and 4a of 123–124° is not affected by the quaternarization reaction of the phosphorus atom, however, we observed a significant opening of the C1−N1−P1 bond angle up to 126° (Table 2). In marked contrast to the structure of 1a which shows a strong localization of the >C1=N1− double bond in the formamidine pattern, the structural parameters of 2b and 4a suggest that the N-phosphonio formamidine derivatives 2a,b, 3b and 4a,b are best described by the combination of the mesomeric N-phosphonio formamidine A1 and iminium phosphazene A4 forms depicted in Fig. 3.
Molecular structure of 2b. Hydrogen atoms have been omitted for clarity except for the formamidine hydrogen atom H1.
Molecular structure of 4a. Hydrogen atoms have been omitted for clarity except for the formamidine hydrogen atom H1.
Selected bond lengths [Å] and angles [°] for compounds 1a, 2b and 4a.
| 1a | 2b | 4a | |
| Distances (Å) | |||
| C1–N1 | 1.289 (3) | 1.326 (6) | 1.309 (3) |
| C1–N2 | 1.341 (3) | 1.313 (6) | 1.311 (3) |
| N1–P1 | 1.697 (2) | 1.609 (4) | 1.605 (2) |
| Angles (°) | |||
| N2–C1–N1 | 123.5 (2) | 122.5 (4) | 123.7 (2) |
| C1–N1–P1 | 115.7 (2) | 126.2 (4) | 126.45 (19) |
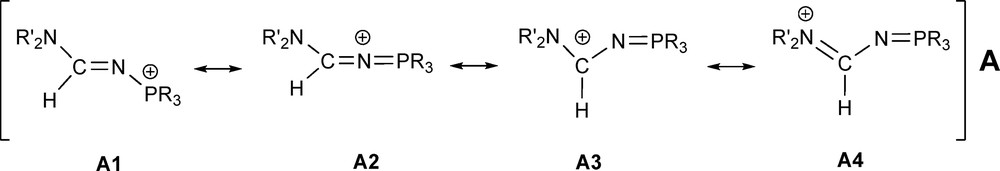
Most representative mesomeric forms of N-phosphonio formamidine derivatives A.
Then, we studied the reactivity of compounds 2a,b towards bases. As expected with methyl phosphonium derivatives, deprotonation reactions on 2a,b in THF at –78 °C with tBuLi led to the formation of the corresponding phosphorus ylides giving rise to signals at δ 31P 40.5 (5a, R = Ph) and 60.7 (5b, R = iPr) ppm (Scheme 2). We were not able to isolate these compounds because of their extreme sensitivity to traces of proton sources to give back the starting phosphonium compounds. The phosphorus ylide 5b was reacted with [(PhCN)2Pd(Cl)2] to form complex 6. The dimeric palladium complex 6 was also prepared in mild conditions starting from 2b, after addition at room temperature of Ag2O as a base and [(PhCN)2Pd(Cl)2] (Scheme 2). The corresponding silver-ylide intermediate was identified by 31P NMR at 70.6 ppm with a set of signals in the 1H NMR spectrum at 0.49 (2JHP = 10.7 Hz) and 8.48 (3JHP = 21.1 Hz) ppm for the ylidic protons P=CH2 and the formamidine proton, respectively. The dimeric palladium complex 6 was fully characterized by mass spectrometry, 1D and 2D 31P, 1H, 13C NMR.
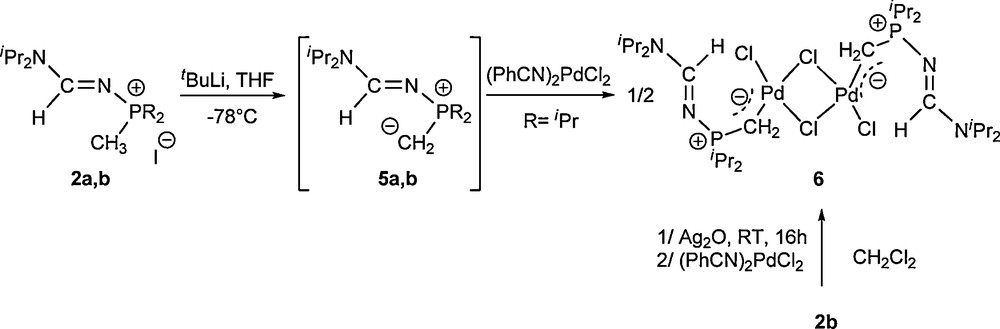
Deprotonation reaction of 2a,b and formation of complex 6.
Mass spectrometric analysis is in full agreement with a dimeric structure for 6. The 1H and 13C NMR spectra of 6 revealed the presence of the proton and the carbon of the formamidine framework >N−C(H)=N− at 8.94 (3JHP = 19.8 Hz) and 159.4 (2JCP = 2.5 Hz) ppm, respectively. In addition to the signals corresponding to the isopropyl substituents connected to the phosphorus atom, the methylene fragment of the ylidic function P–CH2 appears at 1.74 (2JHP = 6.8 Hz) ppm in the 1H NMR spectrum and at –17.7 (1JCP = 31.7 Hz) ppm in the 13C NMR spectrum, shifted to low frequencies in comparison with the chemical shift of the methyl group in 2b. X-ray quality crystals for 6 were obtained from a CH2Cl2/Et2O solution at 4 °C (Table 3).
Selected bond lengths [Å] and angles [°] for 6.
| Distances (Å) | |||
| C1–N1 | 1.283 (6) | Pd1–Cl1 | 2.2845 (15) |
| C1–N2 | 1.346 (6) | Pd1–Cl2 | 2.3397 (15) |
| N1–P1 | 1.616 (4) | Pd1–Cl2’ | 2.4339 (13) |
| C14–P1 | 1.769 (5) | Pd1’–Cl2 | 2.4338 (13) |
| Pd1–C14 | 2.004 (5) | ||
| Angles (°) | |||
| N2–C1–N1 | 121.3 (5) | C14–Pd1–Cl2 | 90.18 (15) |
| C1–N1–P1 | 127.0 (4) | C14–Pd1–Cl2’ | 175.71 (14) |
| N1–P1–C8 | 104.6 (2) | Cl2–Pd1–Cl1 | 178.93 (6) |
| P1–C14–Pd1 | 119.4 (3) | Cl1–Pd1–Cl2’ | 92.99 (5) |
| C14–Pd1–Cl1 | 89.48 (15) | Cl2–Pd1–Cl2’ | 87.29 (5) |
The single crystal X-ray study confirmed the dimeric structure of the ylide complex 6 (Fig. 4). The palladium atoms adopt a square planar geometry. The C14−Pd1−Cl2 and Cl1–Pd1–Cl2’ bond angles of, respectively, 90.2 and 93.0° are very similar. The Pd1−Cl2 bond length (2.3397 Å) is slightly longer than the Pd1−Cl1 bond length (2.2845 Å). This slight distortion is not detected in solution as a single set of signals has been observed for the P−CH2 fragment in the 31P, 1H, and 13C NMR spectra. The P1−C14 bond length of 1.769 Å is comparable with the value recorded in the starting compound 2b for the phosphorus−methyl bond (1.782 Å). The Pd1–C14 bond length of 2.004 Å is one of the shortest recorded to date compared to those generally reported in the literature which are in the average range of 2.03 and 2.19 Å [14]. The structural characteristic of the formamidine pattern >N2−C1(H)=N1− regarding the strong localization of the C1=N1 double bond of 1.283 Å with a significative difference of 0.063 Å between the C1–N1 and C1–N2 bond lengths resembles to the one observed for the N–phosphino formamidine 1a but the C1–N1–P1 bond angle of 126.2° in 6 is typical of tetracoordinated N-phosphorus formamidine derivatives.
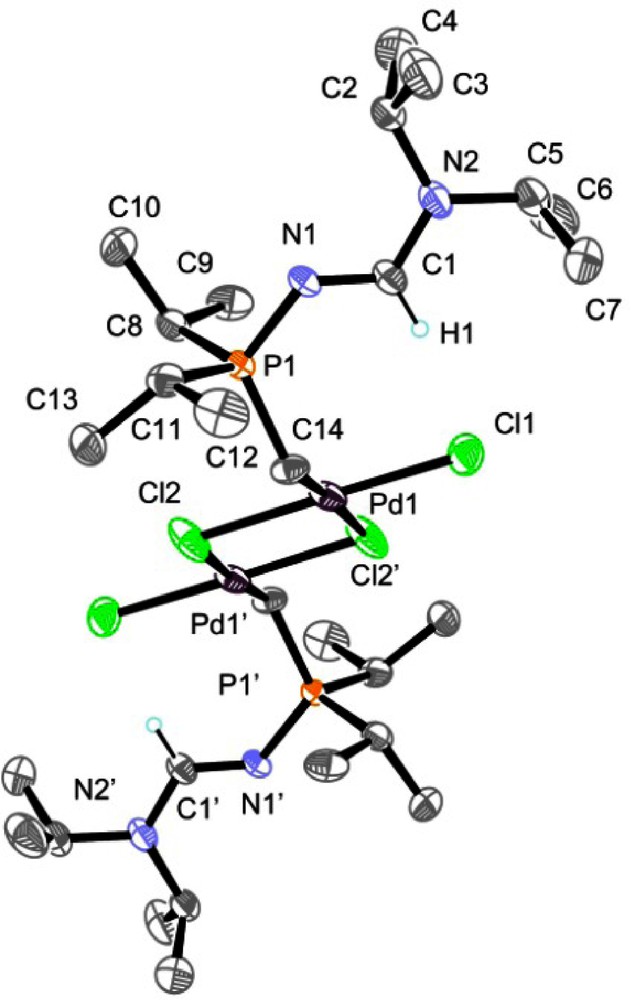
Molecular structure of 6. Hydrogen atoms have been omitted for clarity except for the formamidine hydrogen atom H1.
N-phosphonio formamidines of the general structure [R’2N–C(H)=N–PR3]+X− as 3b (R = iPr) and 4a (R = Ph) should be good precursors to prepare the corresponding amino-iminophosphorane carbene derivatives R’2N−C−N=PR3 [15]. A large variety of bases such as tBuOK, iPr2NLi (LDA), tBuLi, NaH, (Me3Si)2NM (M = K or Li) and mesityllithium (MesLi) have been tested on 3b without any success. Addition at –78 °C of amides MN(SiMe3)2 (M = K or Li) to 4a gave by 31P NMR a signal at –4.0 ppm corresponding to PPh3. The 1H NMR spectrum displayed a set of signals at 1.27 (d, 3JHH = 6.5 Hz) and 3.23 (h, 3JHH = 6.5 Hz) ppm corresponding to diisopropylcyanamide iPr2N−CN (Scheme 3).
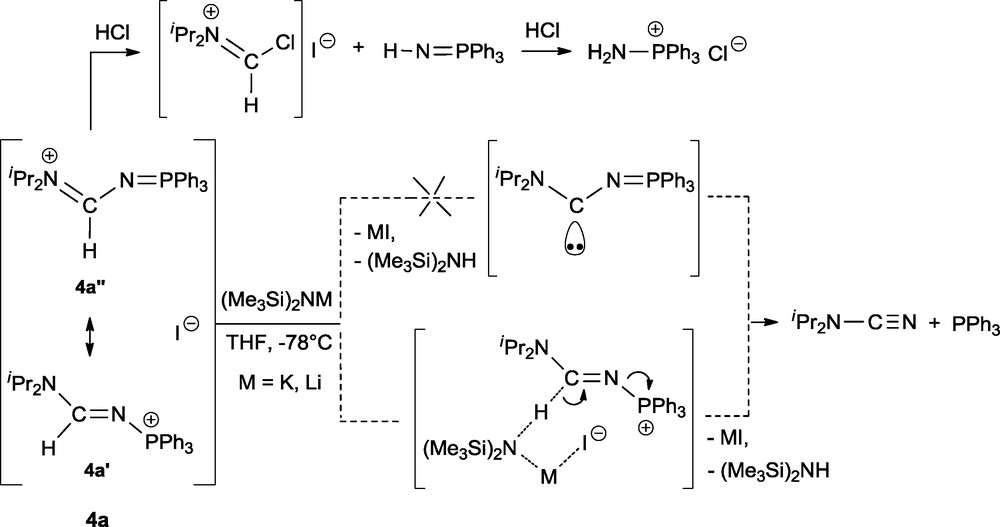
Deprotonation reaction of the N-phosphonio formamidine 4a.
Identification of PPh3 and iPr2N−CN after deprotonation of 4a and elimination of MI (M = K, Li) cannot be rationalized via the transient formation of the amino-iminophosphorane carbene iPr2N−C−N=PPh3. A concerted mechanism should be invoked in this reaction which induces the abstraction of the proton of the formamidine function with concomittant cleavage of the phosphorus-nitrogen bond. The same reaction recorded in the presence of trapping reagents confirmed that the carbene iPr2N−C−N=PPh3 does not form during the deprotonation reaction of 4a. It is interesting to note that the proposed mechanism for the formation of PPh3 and iPr2N−CN in the deprotonation reactions of 4a involves the mesomeric phosphonium form 4a’. In marked contrast, formation of [Ph3P−NH2]Cl [16] as the major phosphorus product after addition of HCl to 4a can be reasonably rationalyzed via the protonation of the basic nitrogen site of the iminophosphorane fragment of the iminium mesomeric form 4a” followed by the cleavage of the carbon-nitrogen bond to give Ph3P=N–H and the corresponding stable iminium compound [iPr2N=C(H)Cl]I. Addition of a second equivalent of HCl led to the observed amino phosphonium product [Ph3P−NH2]Cl.
3 Conclusion
We have prepared in good yields a large variety of N-phosphonio formamidine derivatives of the general formula [R”2N−C(H)=N−P(R’)R2]+X−. The data recorded in solution and the structural parameters of the X-ray analysis revealed that these compounds are best described by the combination of the mesomeric N-phosphonio formamidine and iminium phosphazene forms.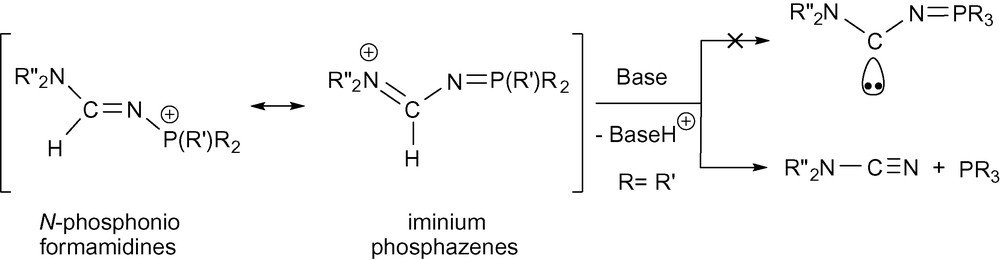
Deprotonation reactions with tBuLi on the N-phosphonio formamidines [iPr2N−C(H)=N−P(CH3)R2]+X− (R = Ph, iPr) led to the formation of the corresponding phosphorus ylides iPr2N−C(H)=N−P(CH2)R2. The phosphorus ylide iPr2N−C(H)=N−P(CH2)iPr2 was reacted with [(PhCN)2Pd(Cl)2] to give the dimeric complex [(iPr2N−C(H)=N−P(CH2)iPr2)Pd(Cl)(μ-Cl)]2 structurally characterized by X-ray analysis. The reactivity of different organic and inorganic bases on [iPr2N−C(H)=N−PR3]+X− (R = Ph, iPr) did not lead to the corresponding carbene derivatives iPr2N−C−N=PR3. Instead, with R = Ph, the deprotonation reaction occurred via an intramolecular rearrangement to give the cyanamide compound iPr2N−C≡N and PPh3. This new class of N-phosphonio compounds [R”2N−C(H)=N−P(R’)R2]+X− will be evaluated as organocatalysts in different organic reactions.
4 Experimental section
All reactions were conducted under an inert atmosphere of dry argon using standard Schlenk-line techniques. Solvents were dried and degassed by standard methods before use. NMR spectra were recorded on a Bruker AV 500, AV 300, DPX 300 or AC200 spectrometers. Chemicals shifts for 1H and 13C are referenced to residual solvent resonances used as an internal standard and reported relative to SiMe4. 31P and 15N NMR chemical shifts are reported relative to external aqueous 85% H3PO4 (31P) and CH3NO2 (15N) respectively. Melting points were obtained using an Electrothermal Digital Melting Point apparatus and are uncorrected. Mass spectra were recorded on a TSQ7000 Thermo Electron mass spectrometer.
4.1 Preparation of iPr2N–C(H)=N–PPh2(Me)+,I− (2a)
MeI (0.19 mL, 3.05 mmol) was added dropwise to a solution of N-phosphino formamidine (1a) (0.95 g, 3.05 mmol) in CH2Cl2 (10 mL) at –78 °C under argon. The reaction mixture was stirred for 5 minutes at room temperature. The solvent was removed under vacuum, the resulting white powder was washed with pentane (3 × 15 mL). Yield: 90% (1.25 g). M.p. 140−142 °C. FT-IR: ν 1612 cm−1. 31P{1H} NMR (121.5 MHz, CDCl3): δ 32.2 (s) ppm. 1H NMR (200.1 MHz, CD2Cl2): δ 1.36 (d, 3JHH = 6.8 Hz, 6H, NCHCH3), 1.47 (d, 3JHH = 6.9 Hz, 6H, NCHCH3), 2.74 (d, 2JHP = 13.2 Hz, 3H, PCH3), 4.04 (h, 3JHH = 6.8 Hz, 1H, NCHCH3), 4.50 (h, 3JHH = 6.9 Hz, 1H, NCHCH3), 7.26–7.37 (m, 2H, HPh), 7.65–7.71 (m, 4H, HPh), 7.75–7.86 (m, 4H, HPh), 8.25 (d, 3JHP = 21.8 Hz, 1H, CH=N) ppm. 13C{1H} NMR (50.3 MHz, CD2Cl2): δ 14.2 (d, 1JCP = 63.3 Hz, PCH3), 20.0 (s, NCHCH3), 22.9 (s, NCHCH3), 47.4 (s, NCHCH3), 48.2 (s, NCHCH3), 125.6 (d, 1JCP = 103.4 Hz, i-PCPh), 130.1 (d, JCP = 12.8 Hz, CHPh), 132,1 (d, JCP = 10.6 Hz, CHPh), 134.4 (d, 4JCP = 3.1 Hz, p-CHPh), 159.3 (d, 2JCP = 7.7 Hz, CH=N) ppm. C20H28IN2P (454.10): calcd. C 52.87, H 6.21, N 6.17; found C 53.62, H 6.15, N 5.95. MS m/z: 327 [M – I]+.
4.2 Preparation of iPr2N–C(H)=N–PiPr2(Me)+,I− (2b)
MeI (0.19 mL, 3.05 mmol) was added dropwise to a solution of N−phosphino formamidine (1b) (0.75 g, 3.05 mmol) in CH2Cl2 (10 mL) at –78 °C under argon. The reaction mixture was stirred for 5 minutes at room temperature. The solvent was removed under vacuum, the resulting white powder was washed with pentane (3 × 15 mL). Yield: 82% (0.97 g). M.p. 124−126 °C. FT-IR: ν 1616 cm−1. 31P{1H} NMR (121.5 MHz, CDCl3): δ 56.6 (s) ppm. 1H NMR (200.1 MHz, CD2Cl2): δ 1.19 (dd, 3JHH = 7.1 Hz, 3JHP = 16.5 Hz, 6H, PCHCH3), 1.21 (dd, 3JHH = 7.1 Hz, 3JHP = 16.5 Hz, 6H, PCHCH3), 1.29 (d, 3JHH = 6.9 Hz, 6H, NCHCH3), 1.32 (d, 3JHH = 6.8 Hz, 6H, NCHCH3), 1.98 (d, 2JHP = 11.4 Hz, 3H, PCH3), 2.42 (hd, 3JHH = 7.1 Hz, 2JHP = 10.3 Hz, 2H, PCHCH3), 4.09 (h, 3JHH = 6.9 Hz, 1H, NCHCH3), 4.16 (h, 3JHH = 6.9 Hz, 1H, NCHCH3), 8.41 (d, 3JHP = 19.7 Hz, CH=N) ppm. 13C{1H} NMR (50.3 MHz, CD2Cl2): δ 4.1 (d, 1JCP = 46.1 Hz, PCH3), 15.4 (d, 2JCP = 3.9 Hz, PCHCH3), 19.7 (s, NCHCH3), 22.5 (s, NCHCH3), 24.3 (d, 1JCP = 66.3 Hz, PCHCH3), 47.2 (s, NCHCH3), 52.2 (s, NCHCH3), 159.7 (d, 2JCP = 3.4 Hz, CH=N) ppm. NMR 15N{1H} (40.6 MHz, d8-toluene): δ = –216.9 (s, NiPr2), –239.6 (d, 1JNP = 37.1 Hz, C=N–P) ppm. C14H32IN2P (386.14): calcd. C 43.53, H 8.35, N 7.25; found C 43.86, H 8.72, N 7.02 MS m/z: 259 [M – I]+.
4.3 Preparation of iPr2N–C(H)=N–PiPr3+,Br− (3b)
N−phosphino formamidine (1b) (0.91 g, 3.73 mmol) was dissolved in 2-bromopropane (5 mL, 53.26 mmol). The reaction mixture was heated at reflux for 18 hours. 2-bromopropane was removed under vacuum, the resulting white powder was washed with pentane (3 × 15 mL). Yield: 86% (1.18 g). M.p. 115−117 °C. FT-IR: ν 1597 cm−1. 31P{1H} NMR (121.5 MHz, CDCl3): δ 56.0 (s) ppm. NMR 1H (300.1 MHz, CDCl3): δ 1.27 (dd, 3JHH = 7.2 Hz, 3JHP = 15.4 Hz, 18H, PCHCH3), 1.28 (d, 3JHH = 6.9 Hz, 6H, NCHCH3), 1.36 (d, 3JHH = 6.9 Hz, 6H, NCHCH3), 2.99 (hd, 3JHH = 7.2 Hz, 2JHP = 11.7 Hz, 2H, PCHCH3), 4.13 (h, 3JHH = 6.9 Hz, 1H, NCHCH3), 4.25 (h, 3JHH = 6.9 Hz, 1H, NCHCH3), 8.74 (d, 3JHP = 17.1 Hz, CH=N) ppm. 13C{1H} NMR (75.5 MHz, CDCl3): δ = 16.6 (d, 2JCP = 2.9 Hz, PCHCH3), 19.6 (s, NCHCH3), 22.6 (s, NCHCH3), 23.2 (d, 1JCP = 54.1 Hz, PCHCH3), 46.4 (s, NCHCH3), 51.3 (s, NCHCH3), 160.3 (d, 2JCP = 4.5 Hz, HC=N) ppm. 15N{1H} NMR (50.7 MHz, CDCl3): δ = –215.6 (d, 3JNP = 9.4 Hz, NiPr2), –245.8 (d, 1JNP = 37.7 Hz, C=N–P) ppm. C16H36BrN2P (366.18): calcd. C 52.31; H 9.88; N 7.63; found C 52.58; H 9.95; N 7.35. DCI MS (CH4) m/z: 287 [M–Br]+.
4.4 Preparation of iPr2N–C(H)=N–PPh3+,I− (4a)
PhI (1.680 mL, 14.99 mmol) and Pd(OAC)2 (0.067 g, 0.30 mmol) were added to a solution of N-phosphino formamidine 1a (3.0 mmol) in toluene (10 mL). The reaction mixture was heated at reflux for 18 hours. The solvent was removed under vacuum, the resulting white powder was washed with pentane (3 × 15 mL). Yield: 80% (1.24 g). M.p. 170−172 °C. FT-IR: ν 1603 cm−1. 31P{1H} NMR (121.5 MHz; CDCl3): δ 30.3 (s) ppm. 1H NMR (300.1 MHz, CDCl3): δ 1.32 (d, 3JHH = 6.9 Hz, 6H, NCHCH3), 1.55 (d, 3JHH = 6.9 Hz, 6H, NCHCH3), 4.13 (h, 3JHH = 6.9 Hz, 1H, NCHCH3), 4.44 (h, 3JHH = 6.9 Hz, 1H, NCHCH3), 7.64–7.82 (m, 15H, HPh), 7.88 (d, 3JHP = 21.0 Hz, 1H, HC=N) ppm. 13C{1H} NMR (75.5 MHz, CDCl3): δ 19.9 (s, NCHCH3), 23.1 (s, NCHCH3), 48.5 (s, NCHCH3), 51.8 (s, NCHCH3), 122.7 (d, 1JCP = 101.9 Hz, i-PCPh), 130.1 (d, JCP = 12.8 Hz, CHPh), 132.8 (d, JCP = 10.6 Hz, CHPh), 134.7 (d, 4JCP = 2.3 Hz, p-CHPh), 157.8 (d, 2JCP = 6.8 Hz, HC=N) ppm. 15N{1H} NMR (50.7 MHz, CD2Cl2): δ –211.9 (d, 3JNP = 12.5, NiPr2), –235.4 (d, 1JNP = 27.8 Hz, C=N–P) ppm. C25H30IN2P (516.12): calcd. C 58.15; H 5.86; N 5.42; found C 58.46; H 5.99; N 5.23. DCI MS (CH4) m/z: 389 [M–I]+.
4.5 Preparation of iPr2N–C(H)=N–PiPr2(Ph)+,I− (4b)
PhI (1.680 mL, 14.99 mmol) and Pd(OAC)2 (0.067 g, 0.30 mmol) were added to a solution of N-phosphino formamidine 1b (3.0 mmol) in toluene (10 mL). The reaction mixture was heated at reflux for 18 hours. The solvent was removed under vacuum, the resulting white powder was washed with pentane (3 × 15 mL). Yield: 28% (0.38 g). M.p. 147−149 °C. FT-IR: ν 1600 cm−1. 31P{1H} NMR (121.5 MHz, CDCl3): δ 51.9 (s) ppm. 1H NMR (300.1 MHz, CDCl3): δ 1.17 (dd, 3JHH = 6.9 Hz, 3JHP = 16.8 Hz, 6H, PCHCH3), 1.23 (dd, 3JHH = 6.9 Hz, 3JHP = 16.8 Hz, 6H, PCHCH3), 1.45 (d, 3JHH = 6.6 Hz, 6H, NCHCH3), 1.46 (d, 3JHH = 6.9 Hz, 6H, NCHCH3), 3.52 (m, 2H, PCHCH3), 4.31 (m, 1H, NCHCH3), 4.40 (m, 1H, NCHCH3), 7.65–7.74 (m, 5H, HPh), 8.75 (d, 3JHP = 17.4 Hz, 1H, HC=N) ppm. 13C {1H} NMR (75.5 MHz, CDCl3): δ 15.5 (d, 2JCP = 2.9 Hz, PCHCH3), 15.7 (d, 2JCP = 2.7 Hz, PCHCH3), 19.8 (s, NCHCH3), 22.7 (s, NCHCH3), 22.7 (d, 1JCP = 5.5 Hz, PCHCH3), 23.4 (d, 1JCP = 4.3 Hz, PCHCH3), 47.0 (s, NCHCH3), 52.0 (s, NCHCH3), 120.9 (d, 1JCP = 97.5 Hz, i-PCPh), 129.4 (d, JCP = 11.5 Hz, CHPh), 132.1 (d, JCP = 8.2 Hz, CHPh), 133.4 (d, 4JCP = 2.7 Hz, p-CHPh), 160.1 (d, 2JCP = 6.4 Hz, HC=N) ppm. 15N{1H} NMR (50.7 MHz, CD2Cl2): δ –213.4 (d, 3JNP = 9.0, NiPr2), –250.5 (d, 1JNP = 32.5 Hz, C=N–P) ppm. C19H34IN2P (448.15): calcd. C. 50.90, H 7.64, N 6.25; found C. 51.44, H 7.89, N 6.12. DCI MS (CH4) m/z: 321 [M–I]+.
4.6 Preparation of complex [iPr2N–C(H)=N–PiPr2(CH2)PdCl2]2 (6)
Ag2O (0.512 g, 2.21 mmol) was added to a solution of phosiumfam 2b (0.854 g, 2.21 mmol) in CH2Cl2 (30 mL) at room temperature. After 16 hours stirring, (PhCN)2PdCl2 (0.424 g, 1.10 mmol) was added to the reaction mixture, which was stirred for 24 hours. The solvent was removed under vaccuum and the crude product was purified on column chromatography using Et2O as eluent. Complex 6 was obtained as a red powder which was recrystalized from a CH2Cl2/Et2O solution at 4 °C. Yield: 25% (0.24 g). 31P{1H} NMR (121.5 MHz, CDCl3): δ 64.6 (s) ppm. 1H NMR (300.1 MHz, CDCl3): δ 1.24 (dd, 3JHH = 6.9 Hz, 3JHP = 11.4, 6H, PCHCH3), 1.30 (d, 3JHH = 6.9 Hz, 6H, NCHCH3), 1.41 (d, 3JHH = 6.9 Hz, 6H, NCHCH3), 1.46 (dd, 3JHH = 7.2 Hz, 3JHP = 11.4 Hz, 6H, PCHCH3), 1.74 (d, 2JHP = 6.8 Hz, 2H, PCH2), 2.45 (h d, 3JHH = 7.2 Hz, 2JHP = 11.9 Hz, 2H, PCHCH3), 3.79 (h, 3JHH = 6.9 Hz, 1H, NCHCH3), 4.35 (h, 3JHH = 6.9 Hz, 1H, NCHCH3), 8.94 (d, 3JHP = 19.8 Hz, CH=N) ppm. 13C{1H} NMR (75.5 MHz, CDCl3): δ –17.7 (d, 1JCP = 31.7 Hz, PCH2), 15.8 (d, 2JCP = 3.5 Hz, PCHCH3), 16.5 (s, PCHCH3), 19.7 (s, NCHCH3), 23.2 (s, NCHCH3), 25.0 (d, 1JCP = 65.4 Hz, PCHCH3), 46.1 (s, NCHCH3), 50.1 (s, NCHCH3), 159.4 (d, 2JCP = 2.5 Hz, HC=N) ppm. DCI MS (NH3) m/z: 888 [M + NH3]+.
5 X-ray analysis
Data of compounds 4a and 6 were collected at low temperature (180 K) on an Xcalibur Oxford Diffraction diffractometer using a graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) and equipped with an Oxford Instrument Cooler Device. Data of compound 2b were collected at low temperature (180 K) on a IPDS STOE diffractometer using a graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) and equipped with an Oxford Cryosystems Cryostream Cooler Device. The final unit cell parameters have been obtained by means of a least-squares refinement. The structures have been solved by Direct Methods using SIR92 [17], and refined by means of least-squares procedures on a F2 with the aid of the program SHELXL97 [18] included in the softwares package WinGX version 1.63 [19]. The Atomic Scattering Factors were taken from International tables for X-Ray Crystallography [20]. All hydrogens atoms were geometrically placed and refined by using a riding model. All non-hydrogens atoms were anisotropically refined, and in the last cycles of refinement a weighting scheme was used, where weights are calculated from the following formula: w = 1/[σ2(Fo2) + (aP)2 + bP] where P = (Fo2+2Fc2)/3. Drawing of molecule are performed with the program ORTEP32 [21] with 30% probability displacement ellipsoids for non-hydrogen atoms.
Acknowledgments
We thank the CNRS for financial support. T.D.L. acknowledges the Agence universitaire de la francophonie (AUF) for a PhD fellowship.
1 The isolated yield of 4b (28%) was not optimized. According to 31P NMR, the product was formed in 75% yield.