

Reactions which generate cyclic acetals are important for the protection of carbonyl groups in organic synthesis [1] as well as for the protection of alcohol groups in carbohydrates [2]. Cyclic acetals are equally important for the generation of chiral auxiliaries for asymmetric induction [3] and for the production of polymers, pharmaceuticals, cosmetics and fragrances [4–6]. The development of new methods and the modification of existing ones for making acetals are therefore considered as an important challenge. Recently, it has been described that cyclic acetals with five to eight membered rings can be obtained by reactions between alkynes and diols catalysed by cationic gold (I) catalyst [7]. Epoxides can be converted directly to cyclic acetals, with only five membered rings, called acetonides, in the presence of acetone and catalysed by a Lewis acid [8]. Classical methods to obtain cyclic acetals involve the reaction of an aldehyde or a ketone with an alcohol, catalysed by toluenesulfonic acid, with azeotropic removal of water or transacetalization reactions. When the ketone is not stable, this procedure involves a large excess of reactant and tedious work-up procedures [1,9]. More convenient and useful methods, under mild conditions, for the production of cyclic acetals of various carbonyls compounds in excellent yields were recently reported, some were using catalytic amounts of tetrabutylammonium tribromide [10] or ZrCl4 [11] in the presence of trialkyl orthoformate, and others were using silylated alcohols with catalytic amounts of trimethylsilyl trifluoromethanesulfonate (TMSOTf) as Lewis acid [12].
Recently, it has been shown that ZrCl4 is an efficient catalyst for one-pot protection/deprotection of diols, where 2,2-dimethoxypropane (DMP) is the protecting agent, forming cyclic acetals [13]. However, there is still a scope for further improvement in this field since there is no efficient method yet for the preparation of cyclic acetals directly from carbonyls and diols without the assistance of additives or co-catalysts.
We report here an efficient and easy method for the preparation of cyclic acetals from diols and carbonyls using hexahydrated Fe (III) trichloride as a catalyst (Scheme 1). This method is proved to be useful for a wide range of substrates. A few examples using anhydrous FeCl3 as a catalyst have been reported in carbohydrate chemistry [14].
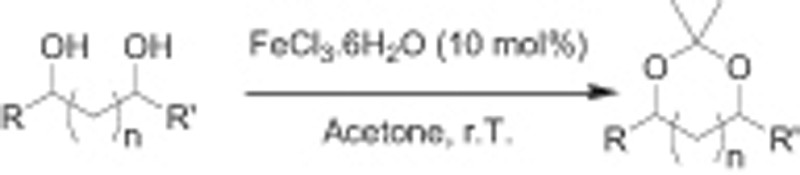
Iron (III) chloride catalyses the transformation of diols into acetonides.
In a preliminary experiment, 1,2-butanediol d5 (Table 1, entry five) was stirred with 10 mol% of hexahydrated FeCl3 and acetone at room temperature for two hours and were subjected to the conditions of the reactions. The results are summarized in Table 1.
FeCl3.6H2O catalyses conversion of diol to acetonides in acetone.
| Entry | Substrate | Conversion (%) | Product | Isolated yield (%) |
| 1 | 100a | 98 | ||
| d1 | 1 | |||
| 2 | 100 | 98 | ||
| d2 | 2 | |||
| 3 | 100 | 98 | ||
| d3 | 3 | |||
| 4 | 74 (100)b | 98 | ||
| d4 | 4 | |||
| 5 | 100 | 98 | ||
| d5 | 5 | |||
| 6 | 100 | 98 | ||
| d6 | 6 | |||
| 65 (78)b | 74 | |||
| 7 | d7 | 7 | ||
| 8 | 50 | 50 | ||
| d8 | 8 | |||
a After 30 minutes.
b After 24 hours.
The conversion of 1,2-diol type d1-d5, α-aromatic substituted and aliphatic diols (Table 1, entries one to five) to the corresponding 1,3-dioxalanes was much faster than the conversion of 1,4-diols type d7-d8 (Table 1, entries seven and eight). This is due to the chelation effect, where diols with closer hydroxyl functions can be coordinated more easily than diols with farther hydroxyl functions. However, the transformation of 1,3-diols such as 2,4-pentanediol was as easy as 1,2-diols type. A diastereomeric mixture of diols d3, d4, d6 and d8 yielded a diastereomeric mixture of acetonides in different ratios as observed by GC-MS and 1H NMR of the reaction products.
In order to further explore the scope of the catalytic process presented here, we have studied a series of reactions involving different combinations of two selected diols with four different carbonyls (Table 2). The diol d1 or d6 was dissolved in THF with one equivalent of carbonyl compound and 10 mol% of hexahydrated FeCl3 and stirred at room temperature for two hours producing the corresponding cyclic acetal1 (Scheme 2).

Iron (III) catalyses the transformation of diols and carbonyls into cyclic acetals.
Reactions between cyclohexanone and d1 as well as d6 were almost completely done with a quantitative yield (> 95 %) (Table 2, entries two and six). However, when 2-phenylpropanone is used, yields were less important than those obtained with cyclohexanone (Table 2, entries three and seven) but much higher than the yields obtained with acetophenone (Table 2, entries one and five).
It seems that this reaction is easier with aliphatic ketones than benzylic and aromatic ones where the conversions were not completed. The reaction of ortho-methylbenzaldehyde with the diol d1 (Table 2, entry four) leads to a good conversion, whereas, complete conversion was obtained with diol d6 (Table 2, entry eight). The cyclic acetals (entries one, three, four, five, seven and eight) obtained in the examples shown in Table 2, exhibit new stereogenic centre except those with cyclohexanone. The use of optically pure diols may induce the stereochemistry of the formed stereogenic centre, this will constitute the object of a future study.
In conclusion, a new method for the preparation of acetonides and cyclic acetals was developed using FeCl3, a very cheap and friendly catalyst. This method uses diols as substrates for the preparation of cyclic acetals of not only five membered rings (as with the use of epoxides) but also for cyclic acetals of six, seven and eight membered rings. Furthermore, this friendly procedure uses ketones, which are commercially available worldwide, and it can be also used for the protection of diols as well as of ketones and aldehydes.
Acknowledgments
We acknowledge Dr. Iman Saad, director of the department of chemistry and biochemistry, and Prof. Bassam Badran for their logistical support.
1 General procedure of the protection of diols with acetone: a 10 mL vial containing a Teflon®-coated stirring bar was charged with FeCl3 (27 mg, 0.1 mmol), acetone (1 mL) and the diol (1 mmol). The resulting solution was stirred at room temperature for two hours. Acetone was removed with a rotary evaporator, and the product was purified on silica gel column chromatography (cyclohexane-AcOEt = 80:20). Its purity (> 98%) was determined by 1H NMR. Conversions were determined by GC coupled with MS. All the final products were isolated and characterized by comparison of their 1H NMR spectra with already reported data (1, [15] 2, [16] 3, [17] 4, [18] 5, [19] 6, [20]). We report here the 1H and 13C NMR for the new compounds: 7, 1H NMR (CDCl3, 300 MHz), d (ppm): 3.85 (m, 1H, O-CH), 3.35 (t, 2H, J = 6.8 Hz), 1.2–1.5 (m, 4H, CH2), 1.25 (s, 6H, O2C(CH3)2, 1.2 (d, 3H, J = 6 Hz). 13C NMR (CDCl3, 75.5 MHz), d (ppm): 108, 73, 67, 34, 27, 25, 21.7. 8 1H NMR (CDCl3, 300 MHz), d (ppm): 3.85 (m, 2H, O-CH), 1.2–1.5 (m, 4H, CH2), 1.25 (s, 6H, O2C(CH3)2), 1.2 (d, 6H, J = 6 Hz). 13C NMR (CDCl3, 75.5 MHz), d (ppm): 105, 74, 32, 28, 22.procedure for the protection of diols/ketones: a 10 mL vial containing a Teflon®-coated stirring bar was charged with FeCl3 (27 mg, 0.1 mmol), THF (1 mL), ketone (1mmol) and diol (1 mmol). The resulting solution was stirred at room temperature for two hours. Then THF was removed and the product was purified on silica gel column chromatography (cyclohexane-AcOEt = 80:20). Its purity (> 98%) was determined by 1H NMR. Conversions were determined by GC-MS. All the final products were isolated and characterized by comparison of their 13C and 1H NMR spectra with already reported data (9, [21] 10, [22] 13, [7] 14, [23]). We report here the 1H and 13C NMR for the new compounds: 11 (tow diastereomers): 1H NMR (CDCl3, 300 MHz), d (ppm): 7.1–7.3 (m, 20H, Ar), 4.95 (dd, 1H, JHH = 6.7 and 8.9 Hz) 4.7 (dd, 1H, JHH = 3.5 and 8.2 Hz), 4.61 (dd, 1H, JHH = 6.7 and 8.4 Hz), 4.13 (dd, 1H, JHH = 0.8 and 6.15 Hz), 4.1 (dd, 1H, JHH = 0.87 and 6.2 Hz), 3.32 (dd, 1H, JHH = 7.9 and 8.8 Hz), 3.02 (s, 2H, CH2-Ar), 2.95 (s, 2H, CH2-Ar), 1.4 (s, 3H, CH3), 1.35 (s, 3H, CH3). 13C NMR (CDCl3, 75.5 MHz), d (ppm): 138, 137, 131, 130.6, 128.5, 128, 127, 126.7, 110, 77, 72, 46, 25. 12 (two diastereomers): 1H NMR (CDCl3, 300 MHz), d (ppm): 7.65 (m, 1H, Ar), 7.59 (m, 1H, Ar), 7.15–7.45 (m, 14H, Ar), 6.3 (s, 1H, -(O)2-CH-Ar), 6.13 (s, 1H, -(O)2-CH-Ar), 5.18 (dd, 2H, JHH = 7.4 and 6.8 Hz), 4.5 (dd, 1H, JHH = 8.3 and 6.33 Hz), 4.0 (dt, 1H, JHH = 7.6 Hz), 3.9 (dt, 1H, JHH = 7 and 0.6 Hz), 3.75 (dt, 1H, JHH = 7.5 Hz), 2.45 (s, 3H, CH3-Ar), 2.44 (s, 3H, CH3-Ar). 13C NMR (CDCl3, 75.5 MHz), d (ppm): 137, 136.6, 135, 130, 128.5, 128, 127, 126.7, 126, 125.7, 103, 79, 72, 17. 15 (tow diastereomers): 1H NMR (CDCl3, 300 MHz), d (ppm): 7–7.4 (m, 10H, Ar), 3.85 (m, 4H, -CH-), 3.07 (s, 2H, -CH2-Ar), 2.82 (s, 2H, -CH2-Ar), 1.05–1.26 (m, 4H, -CH2-), 1.2 (s,3H, CH3), 1.13 (d, 3H, CH3, JHH = 5.8 Hz), 1.1 (s, 3H, CH3), 1 (d, 3H, CH3, JHH = 5.6 Hz). 13C NMR (CDCl3, 75.5 MHz), d (ppm): 139, 128, 127, 126, 101, 73, 45, 40, 25, 22. 16 (two diastereomers): for one diastereomer 1H NMR (CDCl3, 300 MHz), d (ppm): 7.6 (m, 1H, Ar), 7.22 (m, 2H, Ar), 7.12 (m, 1H, Ar), 5.97 (s, 1H, CH-Ar), 4.5 (m, 1H, OCH-), 4.2 (m, 1H, OCH-), 2.4 (CH3-Ar), 1.65 (m, 1H, -HCH-), 1.51 (d, 3H, -CH3, JHH = 6.5 Hz), 1.46 (m, 1H, -HCH-), 1.29 (d, 3H, -CH3, JHH = 6.5 Hz). For other diastereomer 1H NMR (CDCl3, 300 MHz), d (ppm): 7.6 (m, 1H, Ar), 7.22 (m, 2H, Ar), 7.12 (m, 1H, Ar), 5.62 (s, 1H, CH-Ar), 9.96 (m, 2H, OCH2-), 2.4 (CH3-Ar), 1.6 (m, 1H, -HCH-), 1.4–1.42 (m, 1H, -HCH-), 1.30 (d, 3H, -CH3, JHH = 6.5 Hz). 3C NMR (CDCl3, 75.5 MHz), d (ppm): 137, 135, 130, 128, 126, 125.7, 99, 73, 40.5, 21.6, 17.