

1 Introduction
Aptamers were first described in 1990 [1,2] and named with a neologism built up from the Latin expression “aptus” (to fit) and the Greek word “meros” (part) [1]. Aptamers are short single-stranded nucleic acid molecules that can fold into complex, well-defined three-dimensional shapes [1,2]. They form binding pockets and clefts for the specific binding of any given molecule. Targets that are recognized by aptamers can range from metal ions and small chemicals to proteins, viruses or whole cells [3]. These target molecules are bound with high specificity and affinities in the nano- to picomolar, and sometimes even femtomolar range [4]. Aptamers therefore constitute a class of functional nucleic acids with molecular recognition properties comparable to monoclonal antibodies or Fab-fragments.
Nucleic acid aptamers are accessible by enzymatic procedures or by entire chemical synthesis. In vitro, aptamers can be obtained by searching mixtures or so-called “libraries” of many billions of different nucleic acid sequences through a process called in vitro selection or SELEX, an acronym for Systematic Evolution of Ligands by Exponential enrichment. A library of up to 1015–1016 different nucleic acid sequences is generated by automated chemical synthesis. Aptamer libraries display a much higher diversity than any other combinatorial library and thus have an increased probability to successfully isolate a target-binding sequence. The challenge is to select active nucleic acid molecules from this huge diversity. Following combinatorial solid-phase synthesis, the aptamer library is incubated with the immobilized target molecule and tightly binding sequences are separated from unbound ones (Fig. 1). Subsequently, bound aptamers are collected and amplified enzymatically. This library, enriched for target-binding sequences, can then be used for the next selection cycle. Depending on the nature of the target molecule, the selection procedure has to be repeated up to 15 times. Individual aptamer sequences can be obtained from the enriched library by cloning and standard sequencing techniques [5] (Fig. 1). The SELEX process has successfully been used to select aptamers against many different targets but has also some inherent disadvantages. One is the uncertain prediction of the success of the selection. This is mainly due to conformational instability of target proteins, since aptamers recognise defined 3D-shapes [6]. Another problem, reducing in vivo applications so far, is aptamer stability under physiological conditions. To tackle this problem, aptamers can be chemically modified with a variety of functional groups that improve their stability in various biological compartments, enhance the chemical diversity and increase the capacity to recognize their targets [7–10]. Modifications of aptamers are introduced either at the phosphate/ribose backbone or at the nucleobases. Phosphothioates in the DNA backbone enhance the stability against nucleases and the cellular availability. RNA aptamers are most prominently modified at the 2’-ribose, as this position strongly contributes to aptamer stability [11]. Recently the use of locked nucleic acids (LNA) for PCR and in vitro transcription has been described. LNAs contain a methylene ether that bridges the 2’-oxygen atom and the 4’-carbon atom, thereby fixing the ribose in endo conformation [12]. The development of enzymes that allow the adaptation of LNAs for SELEX process will enable the selection of LNA-based aptamers and thereby further expand the chemical diversity and binding capabilities.
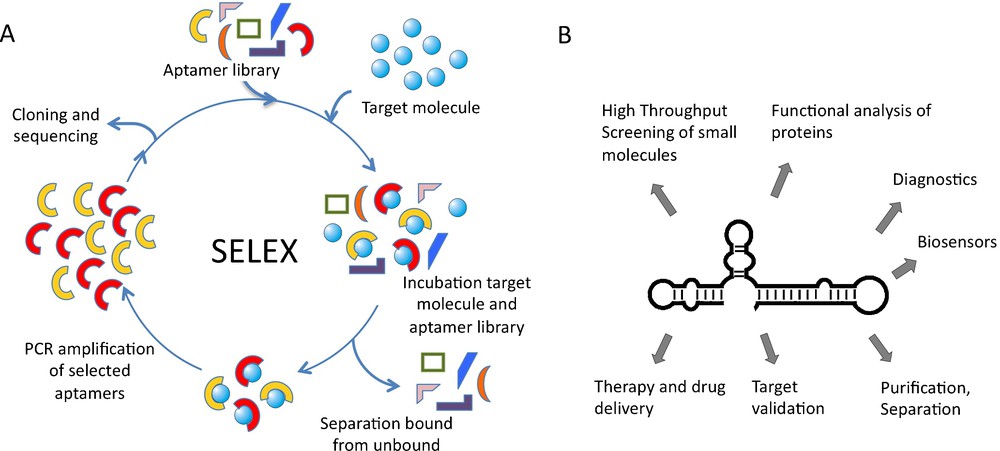
SELEX procedure and aptamer applications. A. Nucleic acid libraries are incubated with the target molecule, binding sequences are separated from unbound and amplified by PCR. Selected sequences can be used in additional cycles. B. Aptamer applications ranging from basic research to medicinal therapy.
Over the last several years, aptamers have become increasingly important in both basic research and biomedical application and are expected to have an even higher impact in the future [5,6,13–15]. Aptamers have been used for various purposes in diagnostics, biosensing, chemistry, and molecular biomedicine. These include the application of aptamers as inhibitors of target molecules, which is particularly important for target validation and functional characterisation in cellculture experiments and in vivo [5]. In addition, aptamers are applied in drug discovery, purification and separation purposes and for method development (Fig. 1). Aptamer-ligand binding is often attended by structural alterations of the aptamer that can be converted into a detectable signal. Through direct coupling of molecular recognition to signal generation, aptamers can be enabled to function as versatile reporters [13,15]. It is also envisioned to utilize aptamers as drugs or as tools for drug-delivery [6,16].
The following chapters address two recent examples from our research, FACS-based SELEX and aptamer displacement, demonstrating how aptamers can be applied in screening procedures. In the final part, we extend our research to nano-engineering based on interlocked, entirely double-stranded DNA architectures.
2 Aptamer displacement
To identify molecules of biological or medicinal interest, our previous and current aim is to develop approaches that combine evolution of biomolecules with small molecule screening. Using the SELEX process, aptamers are isolated in vitro out of the highly diverse nucleic acid libraries by adaptation to external conditions [7,16]. In vitro, aptamers can serve as specific inhibitors of any protein target. While the delivery of aptamers into cells is as straightforward as analogous delivery methods for other functional nucleic acids, particularly siRNAs, the delivery-problem of functional aptamers into tissues or whole organisms is still largely unsolved, hampering their broad therapeutic or clinical application for intracellular targets. This renders aptamer approaches suited only for limited clinical use. Compared to nucleic acid or biopolymer-based inhibitors, small molecules are highly advantageous with respect to cell permeability and chemical diversity. On the other hand, direct screening of small molecules can be tedious and often screening methods need to be tailored to the corresponding drug target [16]. To combine the advantages of small molecules and the aptamer selection process, we developed methods to convert an aptamer-protein complex into a small molecule inhibitor. This is realised in a two-step process. First, an aptamer is identified by very fast and straightforward SELEX procedure providing rapid access to inhibitory molecules the effect of which can be tested in initial cell culture experiments. Second, if a promising effect is observed, small molecule libraries are screened for compounds that displace the aptamer from its target and adopt its inhibitory activity [8–10,17]. By this approach, small molecules with similar inhibitory activities as the parent aptamer can be directly searched for, opening new ways to rapidly identify alternative small molecule target sites in proteins. Using the binding properties of the aptamer, the screen for small molecules is target-independent and broadly applicable. This screening method is particularly attractive when no functional assay is available. In addition, no chemical label of proteins and library compounds is required [5,7,15,18]. In the following, the concept of aptamer displacement is illustrated by two recent examples form our research.
In the first example, we have used an aptamer directed against reverse transcriptase from HIV-1, combined with a hammerhead-ribozyme to yield an allosteric ribozyme that cleaves a fluorescence-labelled oligonucleotide substrate in response to the presence or absence of the target protein [8]. This allosteric ribozyme was applied in a high-throughput screening assay to search for small molecules that bind to the protein in a way that no longer permits its binding to the anti-HIV-1 aptamer portion. Thereby, the ribozyme's ability to cleave the substrate was altered, which could be detected in a change of fluorescence (Fig. 2). This screening assay identified SY-3E4 as a new small molecule inhibitor for HIV-1 RT that was able to inhibit the replication of HIV-1 wild-type and multidrug resistance strains [19].
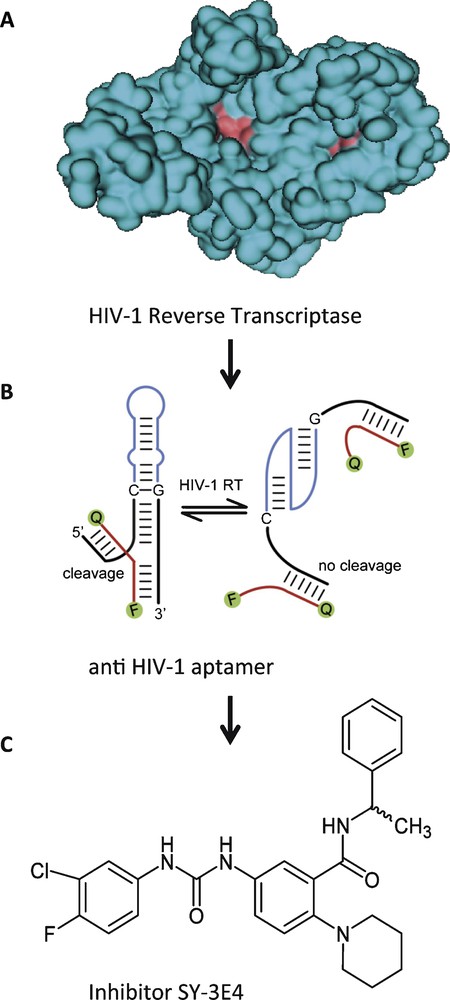
Small molecule SY-3E4 inhibits HIV-1 reverse transcriptase. A. Spacefilling model of HIV-1 RT. B. HIV-1 RT-responsive hammerhead-ribozyme. C. Structure of the selected small molecule inhibitor SY-3E4.
Adapted in parts from [19].
Another example of how aptamer displacement screening can be applied for small molecule screening is the identification of an antagonist of the cytohesin class of small guanosine exchange factors (GEF). GEFs constitute a large and diverse protein family that enables exchange of bound GDP to GTP on small G-proteins and thereby activates them [20,21]. GEF inhibitors are of interest to elucidate the function of this protein family and as therapeutic agents, as their cellular targets are involved in various disease-related signalling networks. At the time when we started the project, the only small molecule that was described as GEF inhibitor was brefeldin A (BFA) [22,23]. A class of BFA-resistant small GEFs for ADP-ribosylation factors (ARFs) are cytohesins. There are four known cytohesins in mammals and in humans, which regulate cytoskeletal organization, integrin and insulin signalling [9,24,25]. Common to all cytohesins is a conserved domain structure: the amino-terminal coiled-coil domain, which is used for interaction with cellular-binding partners, the central Sec7 domain, which contains the GEF function, and the pleckstrin homology (PH) domain, which can regulate the membrane association.
We have identified a series of RNA aptamers that specifically bind the Sec7 domain of cytohesin1 [26]. The selected aptamer M69 inhibits the guanine–nucleotide exchange on ARF1, thereby preventing ARF activation in vitro. M69 is specific for small GEFs, as it shows considerably weaker interaction with members of the class of large GEFs [26]. M69 was used in subsequent studies to establish a small molecule screening for cytohesin inhibitors [9,17,27]. The assay is based on fluorescence-labelled aptamers and the idea to use small organic compounds to displace the aptamer from its bound protein target. The association of the aptamer with its target is detected by fluorescence polarization. Fluorescence polarisation is caused by the rotational diffusion of a molecule. This parameter is inversely proportional to the molecular volume and therefore indirectly proportional to the molecular weight of a fluorescence-labelled aptamer-protein complex. The technique of fluorescence polarization has been used to detect a variety of molecular interactions, like protein-protein, protein-nucleic acid, and receptor-ligand [27]. In our experimental setup, target-binding of the small molecule results in aptamer displacement from the protein of interest and thereby in an increase in fluorescence polarisation (Fig. 3). The method can easily be automated by use of a plate reader and is compatible with high-throughput screening. We screened thousands of different small compounds and identified SecinH3 (Sec7-inhibitor H3) as the most potent cytohesin inhibitor [9]. In various studies, the potential of SecinH3 (Sec7-inhibitor H3) as an indirect inhibitor of ARF1 and ARF6 could be proven [9,28]. With the help of this small compound, the involvement of cytohesins in insulin signalling was shown. In human HepG2 cells and in murine liver cells SecinH3 blocks the transcription of insulin-dependent genes [9]. More recently, we found that the cytohesin ARNO (or cytohesin-2) is also involved in the activation of another class of receptor tyrosine kinases, namely the class of ErbB receptors, including the epidermal growth factor receptor (EGFR), and again SecinH3 and M69 served, among other approaches, as useful chemical biology tools for this purpose [29]. These examples show how, starting from aptamer selections, one can arrive at useful small molecule tools with similar properties as the parent aptamer that can be applied in in vivo model organisms, by using aptamer displacement.
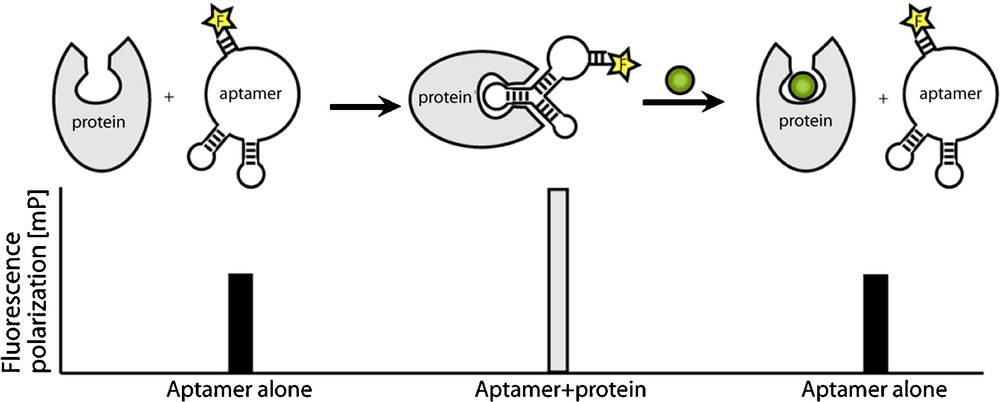
Aptamer displacement screen by fluorescence depolarisation. The unbound aptamer shows low polarisation. Binding to target protein causes increased fluorescence polarisation. If the aptamer is displaced from the protein by a small molecule (green), fluorescence polarisation decreases.
Adapted from [27].
3 FACS SELEX
These and many other examples show that aptamers are useful tools for functionally characterising biomolecules [5]. In addition, aptamers can be used as therapeutics or diagnostic tools, for example to selectively identify malignant cells [6]. Over the past years a substantial amount of research has been devoted to the development of cell-specific aptamers that can discriminate between vital and pathogenic cells [30].
In contrast to the standard SELEX procedure, targeting soluble and purified proteins or extracellular domains of receptors, one can directly employ cells as targets during the cell-SELEX selection process. This approach has the advantage of being able to enrich cell-specific aptamers that target distinct cells or even cell-subpopulations out of complex cell mixtures, without any prior knowledge of the primary target of the aptamer. To facilitate a cell-SELEX experiment, two prerequisites must be fulfilled. First, suitable cells need to be accessible. Many cell types can be obtained as immortalized cell lines or primary cells can be isolated from tissue. Second, cells with reduced membrane integrity need to be eliminated, because these cells unspecifically bind and take up nucleic acids, counteracting the success of the SELEX procedure. So far, two techniques are used to obtain cells for SELEX experiments. Adherent cell lines are separated by repeated washing with buffer and cells grown in suspension are prepared by centrifugation. Both methods have the problem to distinguish between dead and vital cells in the cell suspension obtained. Apoptotic or necrotic cells cannot be separated from vital cells. This is a major problem, since cells with reduced membrane integrity show a high affinity for nucleic acids, which is sequence-independent and leads to the enrichment of unspecific, unwanted aptamer sequences. In addition, unspecific DNA-binding increases the number of selection cycles, making the SELEX procedure inefficient or causes, in the extreme case, the failure of a SELEX experiment.
To circumvent these problems, we have implemented fluorescence-activated cell sorting (FACS) in the selection process [31]. A flow cytometer is used to select a subpopulation from composite mixtures of cultured cells, e.g. dead or vital cells or cells displaying a specific protein on the surface. Cells are separated on the bases of different light-scattering properties or the ability to enzymatically convert cell-permeable dyes into a fluorescent form [32]. Consequently, only the selected cell subpopulation is targeted during the selection process. In this study we used CD19+ Burkitt's lymphoma B-cells as an example to specifically target tumour cells with DNA-aptamers. A composite cell mixture, containing vital as well as dead cells with reduced membrane integrity, was incubated with a single-stranded DNA (ssDNA) library and the dye calcein AM, to visualize an active metabolism. Dead and vital cells were sorted by use of FACS, thereby selecting bound aptamers associated with vital cell phenotype. The flow cytometer therefore simultaneously isolates cell population of interest and removes unbound nucleic acids species [32]. The selected nucleic acids are amplified by PCR. After single-strand displacement, the resulting single-stranded DNA library serves as starting library for the next selection cycle (Fig. 4). After 10 rounds of selection, an aptamer library with enhanced binding to vital CD19+ Burkitt's lymphoma cells could be enriched. Subsequent cloning and sequencing of this library showed several aptamer sequences, out of which one representative clone, C10, was further analysed. C10 DNA-aptamers were conjugated with fluorescein and following incubation the fluorescence intensity distribution of cells was measured [31]. The fluorescence-labelled C10 aptamer showed enhanced binding to vital CD19+ Burkitt's lymphoma B-cells, compared to the starting ssDNA library (Fig. 4). In comparison, a scrambled control DNA sequence showed no binding of B-cell lymphoma. A dissociation constant of 50 nM was determined for the interaction between C10 aptamer and Burkitt‘s lymphoma cells. No interaction of C10 with an acute T-cell leukemia cell line (Jurkat E6) and also no interaction with CD19+ B-cells isolated from healthy volunteers was observed. Taken together, these results show that C10 aptamer binds Burkitt lymphoma B-cells with high affinity and specificity and can distinguish between Burkitt lymphoma B-cells and primary B-cells [31]. Interestingly, the C10 aptamer could be enriched after only 10 cycles of selection. A low number of selection cycles is advantageous, since the aptamer sequences obtained are less prone to selection artefacts than with a higher number of selection cycles used in conventional SELEX experiments [32].
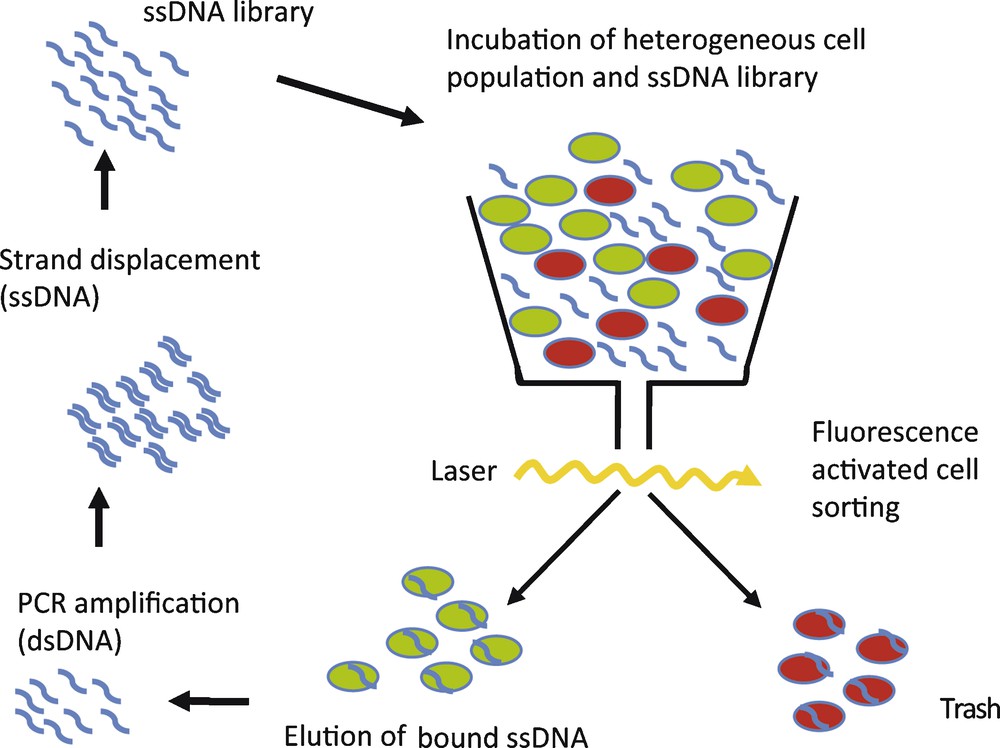
Schematic of FACS SELEX. A heterogeneous cell population, containing dead (red) and vital or different cell types (green), is incubated with single-stranded (ss) aptamer library. Cells are separated by FACS and selected sequences are amplified.
The new FACS SELEX is a fast and robust method to select cell-specific aptamers. It is generally applicable to the screening of single-stranded DNA-libraries and is the method of choice to select an aptamer against cells grown in suspension or primary cell isolates. It could be shown that the combination of FACS technology with the SELEX process increases the efficiency and the success rate of the selection. FACS SELEX is the first approach that identifies and eliminates cells from complex cell mixtures to enrich for cell-specific aptamers [31,32]. Thus, the selection is based on association of aptamers with vital cells, rather than on high affinity binding of a homogeneous target molecule, as in a conventional SELEX experiment. The new technique can be applied to select tumour and cell-specific aptamers that are able to target subpopulations of cells out of complex mixtures. Selected aptamers can distinguish healthy from malignant cells, isolated directly from primary tissues or body fluids. This can be a first step towards the use of high affinity, specific cell-targeting aptamers as individualized diagnostic and biomedical agents complementary to widely used antibodies [32]. In addition, aptamers could be used as precursors for the setup of aptamer displacement screens to find small molecule lead structures, as described in Section 2.
4 DNA nanoarchitectures
Recently we have started to expand our research in the field of modular functional nucleic acids to interlocked DNA architectures, based on circular geometries. In particular, we sought to construct mechanically interlocked double-stranded DNA architectures like rotaxanes and catenanes, in which individual components can be set in motion in a controlled manner. Rotaxanes consist of a dumbbell-shaped molecule threaded through a macrocycle that can move along the axle, trapped by bulky stoppers. Various examples of interlocked rotaxane structures with ingenious functions have been realized in organic synthesis [33]. We have recently reported the design, assembly, and characterization of rotaxanes in which both, the dumbbell-shaped molecule and the macrocycle, are made of dsDNA [34]. These dsDNA-based rotaxanes represent attractive devices for nanorobotics and synthetic biology.
The design of the circular geometries is based on AT-tract secondary structure elements, which cause an intrinsic curvature of the helical axes and therefore allow an efficient synthesis of dsDNA nanorings even if the rings contain single-stranded (ss) gap regions. The gap rings can be used as versatile building blocks in DNA nanotechnology due to their ease of hybridization with a variety of functional oligonucleotide motifs like chemically modified ssDNA [35], RNA hairpins [36], or struts made of dsDNA [37] or Dervan-type polyamides [38].
In the assembly of DNA rotaxanes, these gap rings serve as templates in the threading process [34]. Hybridizing a DNA rod with a single-stranded region of 13 base pairs (bp) and a DNA gap ring with a complementary single-stranded region of 8 bp led to almost quantitative threading (Fig. 5A). Subsequent ligation of simple 168 bp ring stoppers on both sides of the DNA rod produced dsDNA pseudorotaxanes (Fig. 5B). The correct constitution of the interlocked architecture was proven by AFM scans of the purified pseudorotaxanes (Fig. 5C).
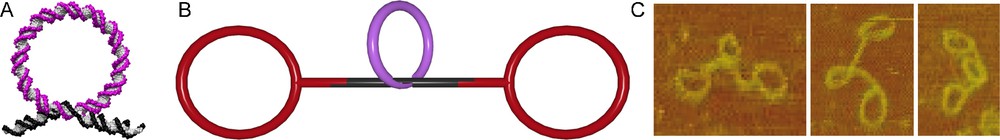
DNA pseudorotaxane. A. DNA rod (black) is threaded through a macrocycle (magenta) by hybridization. B. The DNA pseudorotaxane is assembled from a macrocycle (magenta), a rod (black) and the stoppers (red). C. AFM scans of the DNA pseudorotaxane.
We showed that the macrocycle can be released from the rod by adding short so-called release-oligonucleotides (ROs) that are complementary to the gap sequences. In this case, however, the pseudorotaxane disassembled to the dumbbell and the free macrocycle by a slippage mechanism. Slippage could be prevented, when the single-ring stoppers were substituted by spherical stoppers composed of two intertwined crossover rings (Fig. 6A). With these stoppers, the addition of the ROs led to conversion of the pseudorotaxane into a genuine dsDNA rotaxane (Fig. 6B), the integrity of which was verified by AFM. The free mobility of the macrocycle was demonstrated by fluorescence spectroscopy [34].

Assembly of the dsDNA rotaxane. A. Spherical stopper based on two 168 bp crossover rings. B. Molecular model of the genuine double-stranded DNA rotaxane. C. AFM scans of the rotaxane (scale bar 100 nm).
These dsDNA rotaxanes have the potential to be applied as versatile components in nanomechanics and nanorobotics. Moreover, our broadly applicable and straightforward method for creating interlocked dsDNA architectures opens up new possibilities for constructing DNA-based molecular machines.
Acknowledgements
Our work is supported by the Alexander von Humboldt Foundation, the Deutsche Forschungsgemeinschaft, the European Science Foundation, the Fonds der Chemischen Industrie, the European Research Council, and the Land Northrhine Westphalia. We thank the past and present members of the Famulok lab for their contributions to these projects.