

The research subject of α–galactosylceramides and their behavior as immunostimulants began in 1993–1994. The start came with reports by scientists from the Kirin Pharma labs of anticancer activity in an in vivo mouse assay of a mixture of glycolipid materials isolated from a sponge Agelas mauritanius [1]. The extracts were first shown to be a mixture of α-galactosylceramides 1 named agelasphins. A structure/activity optimization effort led to a synthetic analog 2 named KRN7000 which became the lead compound for studies of its mechanism of action as an immunostimulant [2].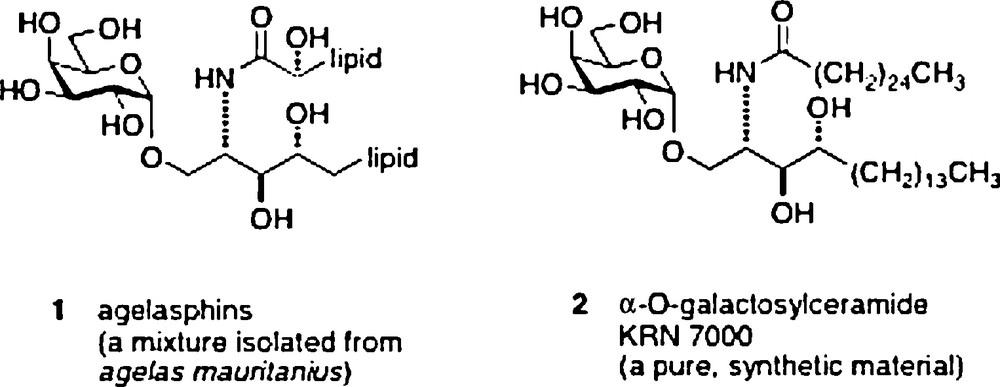
To summarize a large body of work, researchers demonstrated that the two lipid chains of the ligand are complexed by a receptor on the surface of antigen-presenting cells (APC), identified as the CD1d receptor. This receptor has two hydrophobic channels which recognize lipids, and in the specific instance of galactosylceramides, presents the galactose, largely unbound, on the surface of the receptor. This binary complex is then recognized and bound by a receptor on the surface of natural killer T cells (NKT), which binds to the galactose OH's, the phytosphingosine 3’OH and also to surface protein residues of the CD1d receptor. The NKT receptors are very similar in mouse and human and can cross-react with either mouse or human CD1d-ligand complexes. Upon formation of the ternary complex, the two cells displaying the receptors are stimulated to secrete cytokines, IL-12, IL-4 and IFNγ, the first by APC and the last two by NKT. The crystal structure of the ternary complex (Fig. 1) reported in 2007 confirmed the binding model which had been developed based on earlier X-ray structural results with the empty CD1d receptor and with the binary complex [3].
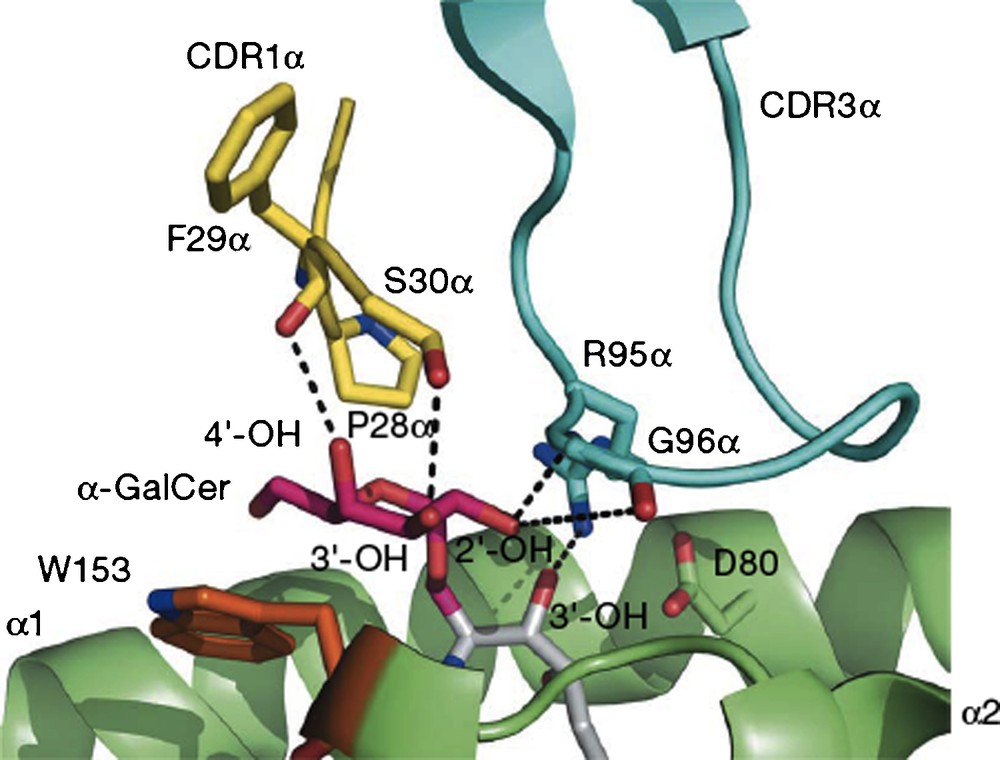
A view of the interface of the proteins CD1d (green) and NKT receptor (blue and yellow) linked by O-galcer (red/gray) (reprinted from [3] with permission).
Contemporary with the basic research on the structure determination of its active complex and on the mechanism of the effect on the immune system by the ligand, a major effort was put forth to find applications of the immunostimulant effect in murine models of diseases found in humans [4]. Parallel to the extensive studies of the immunology effects promoted by KRN7000, several laboratories prepared many dozens of analogs in an attempt to alter the cytokine profiles produced by the parent material when complexed. The analogs can be categorized by:
- • fatty amide chain variation;
- • phytosphingosine chain and functionality variation;
- • substituent variation at the carbohydrate 2, 3, 4 and 6 positions;
- • replacement of the anomeric O linker by other atoms;
- • replacement of the ring O by C. These studies have been thoroughly and very recently reviewed [5].
Our laboratory has been active in the chemical synthesis of C-glycoside analogs (α–C–galcer) of α–galactosylceramide and then providing samples to the immunology community. In the following sections, there will be presented first a very short summary of all the reported syntheses of α–C–galcer with commentary on the challenges faced in the chemistry. Then, a review of the published in vivo results with α–C–galcer assayed in various mouse models of disease will be offered. Lastly, there is a short discussion which surveys the interpretations used to rationalize the observed biological effects.
1 Synthetic chemistry
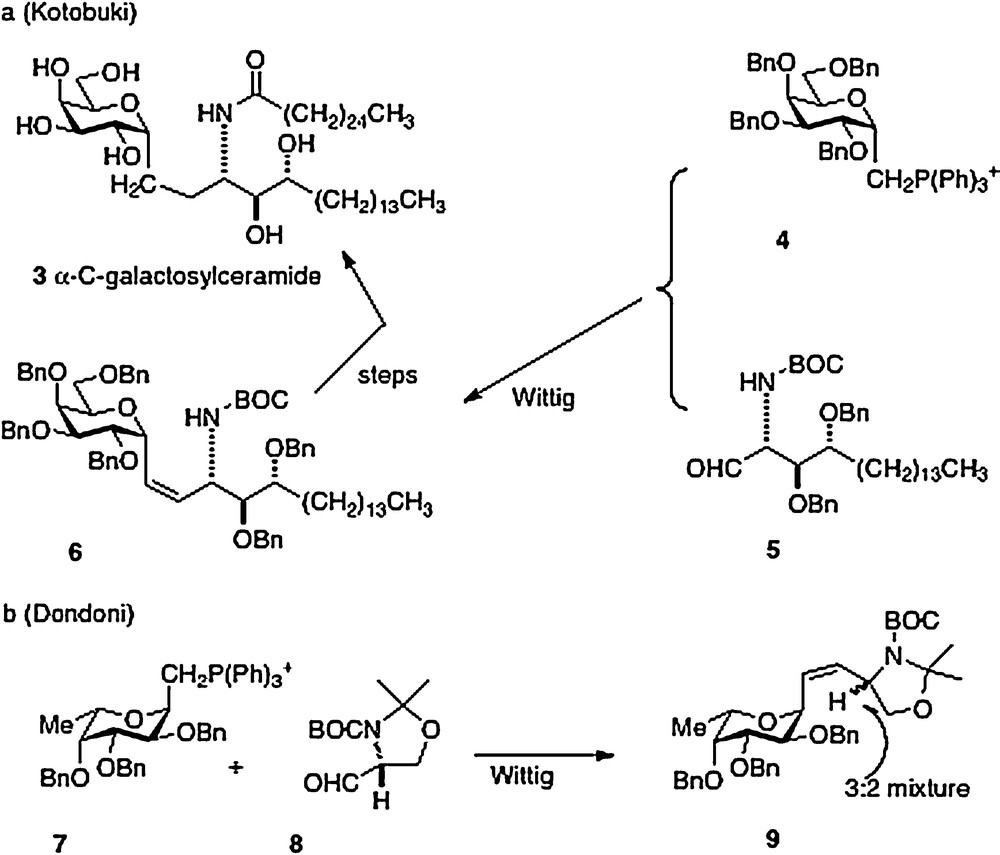
The first synthesis of α–C–galcer 3, by Kotobuki Pharma in 2002, was reported in the patent literature [6]. The key synthetic step is a Wittig reaction between the D-galactosyl triphenylphosphonium ylide 4 and the aldehyde derived from phytosphingosine 5 to afford 6 in low yield. Further straightforward elaboration was reported to produce the desired α–C–galcer (Scheme 1a). It is worthy to note that Dondoni in 2004 [7] described a similar approach with the L-fucosyl Wittig reagent 7 and the aldehyde 8. Analysis revealed that the Wittig conditions had caused epimerization at the amine-bearing carbon presumably at the aldehyde 8 precursor stage, affording a 3:2 inseparable mixture of epimers 9 (Scheme 1b). Then in 2005, Annoura, in his report of a synthesis of a C-galcer analog with a truncated sphingosine chain (vide infra), asserted in a footnote that his group could not reproduce the chemistry described in the Kotobuki patent [8].
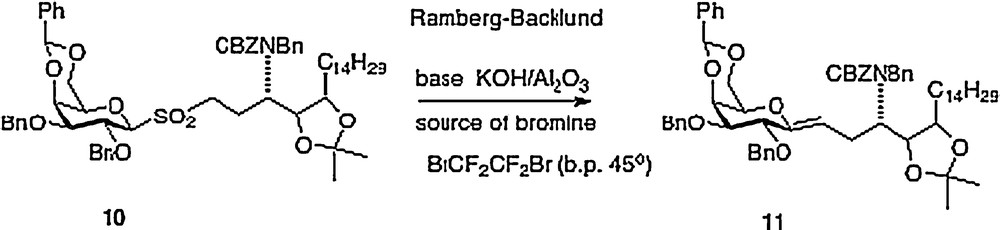
My Hunter group has described four different approaches to α–C–galcer, the first route was published in outline form in 2003 with a more detailed chemistry disclosure following in 2004 [9]. This report featured a key Ramberg-Backlund reaction of sulfone 10 which results in a direct linkage of a protected galactose to a homophytosphingosine affording C–galcer 11 which then requires elaboration to the desired target 3 (Scheme 2).
This route requires more than 20 synthetic steps and would require process development to be practical. The synthesis of the homophytosphingosine element could be shortened by several steps if one were to start with commercially available phytosphingosine. But the key Ramberg-Backlund step has become suspect because the reagent C2Br2F4 is now on the “ozone list” and is no longer commercially available; therefore a further exploration for a suitable reagent would be required. Two reasonably concise syntheses of 3 were then developed to ensure its ready supply for testing once we had recognized its potency (Scheme 3).
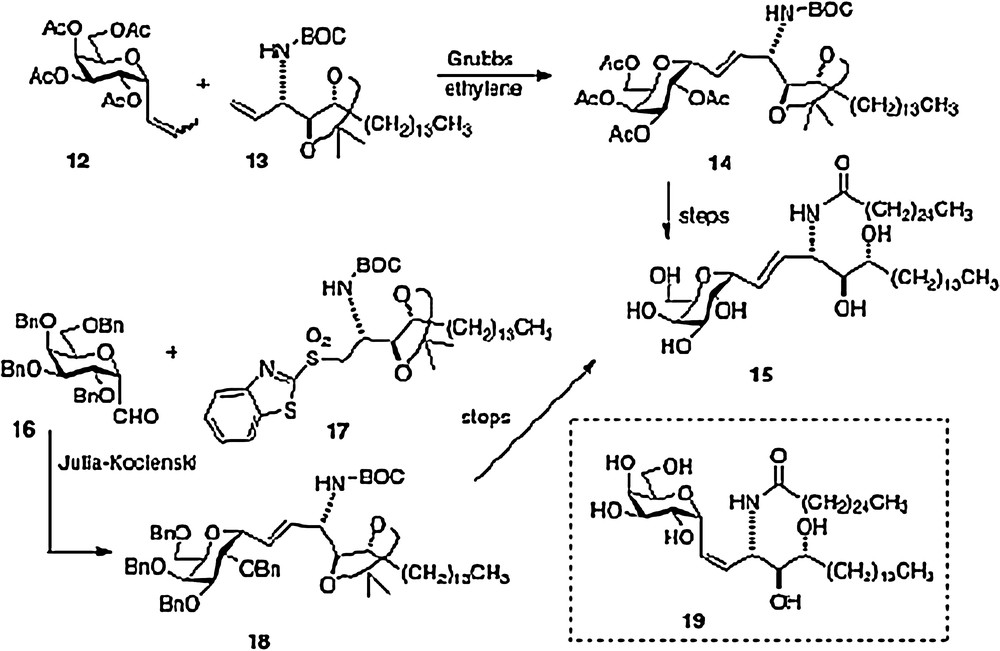
An olefin cross-metathesis between easily prepared α-1-C-propenylgalactose 12 and vinylated phytosphingosine 13 affords alkene 14 which is simply processed to our active α–C–galcer 3 and its alkene analog 15 [10]. The novel feature of this route is the use of ethylene to promote two metatheses in one pot: first C-propenylgalactose to C-vinylgalactose, second-merger of vinylgalactose with vinyl phytosphingosine. This facile scheme needs process development as well because vinyl derivative 13 is currently derived from a precursor aldehyde which is very susceptible to epimerization (see above discussion of the Dondoni route).
The second concise synthesis in our lab uses the Julia-Kocienski reaction as the convergent step, linking galactosyl aldehyde 16 and phytosphingosine sulfone 17 to produce alkene 18 and subsequently either 3 or 15. We were pleasantly surprised to find that aldehyde 16 could survive the basic conditions of the condensation without epimerization or elimination. Using the Charette modification of the Julia-Kocienski chemistry, we were also able to obtain a usable quantity of the Z-analog 19. Thus, we were able to test two alkene C-galcer analogs in addition to the parent C-analog in our bioassays [11].
Our fourth route, published in 2008, was linear in style [12]; thus an epoxide 21, available in 3 steps from aldehyde 20, is ring-opened to afford azide 22. Reduction of the azide and protection of the resulting amide precedes conversion of the primary alcohol of 22 to an aldehyde. Then organometallic chemistry affords a mixture of isomers 23, which, after separation, leads straightforwardly to α–C–galcer 3 and its 4′ epimer (Scheme 4). This route does open the way to diversifying the phytosphingosine chain, but is hampered by its linearity and by problems with getting clean inversion in the epoxide-opening step.
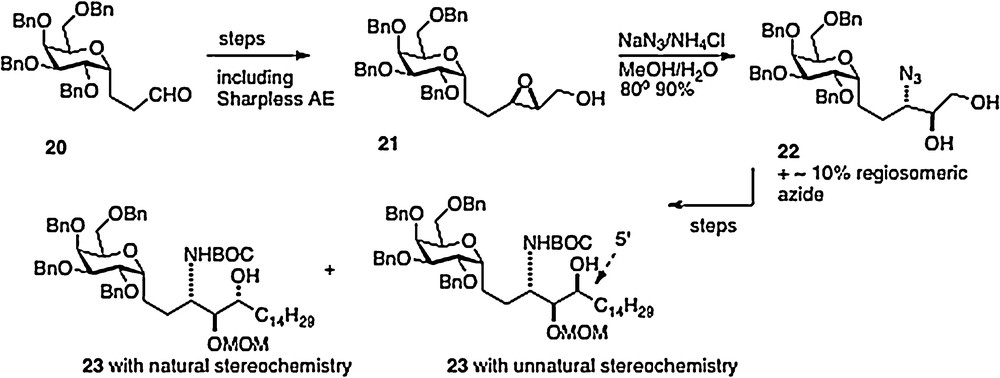
A fifth synthesis of α–C–galcer 3 has been described by Wipf [13]. This work is also linear in approach with nitrogen introduction via organometallic chemistry of sulfone-imine 24, available from precursor aldehyde 20 described above. The product 25 is easily transformed to the target 3.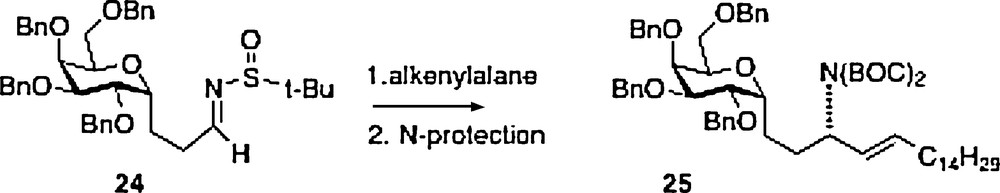
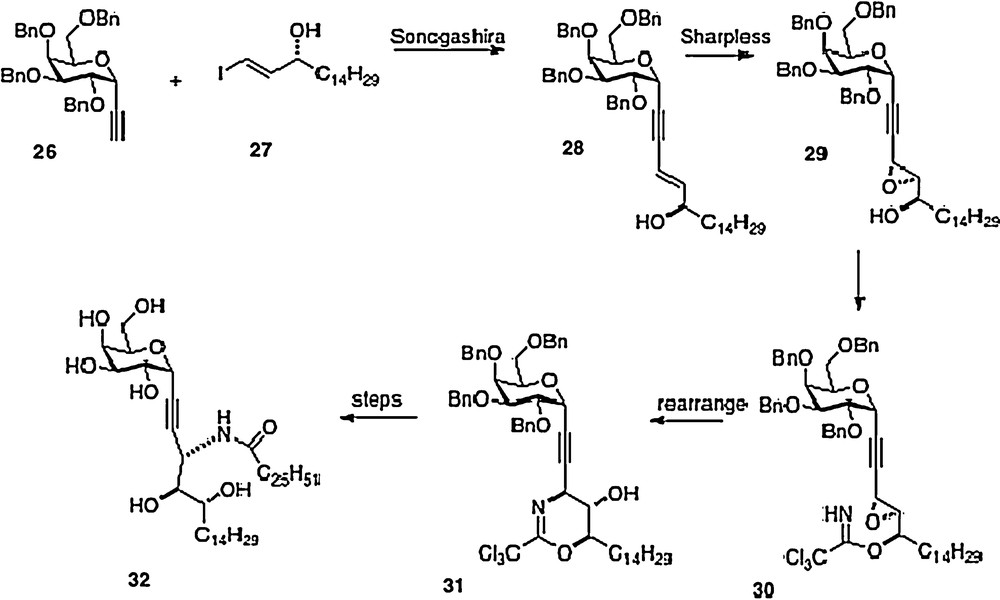
A sixth synthesis has recently been reported by Bittman [14]. Sonogashira coupling of alkyne 26 with iodoalkene 27 affords phytosphingosine precursor 28. Introduction of the remaining oxygen and nitrogen functionality is done via epoxide 29 and trichloroacetimidate 30. This last material is rearranged to 31 which then is converted to alkyne analog 32, or alternatively to 3 (Scheme 5).
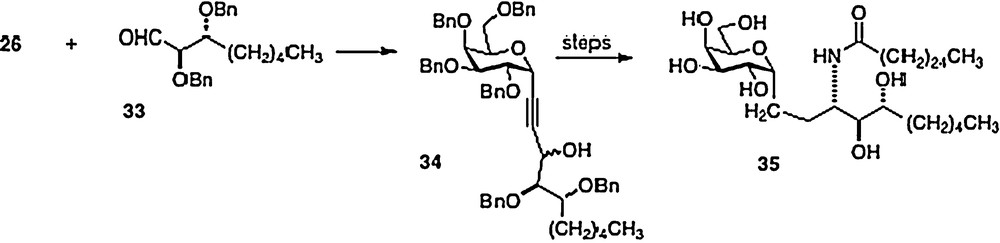
In 2005, Annoura [8] reported the preparation of a truncated phytosphingosine analog of 3. Coupling alkyne 26 directly with aldehyde 33, Annoura obtained alcohol 34 as a mixture of epimers. After separation, the major epimer was simply converted into the truncated analog 35 (Scheme 6). Clearly, any phytosphingosine chain length is accessible via the Wipf, Annoura and Bittman chemistries.
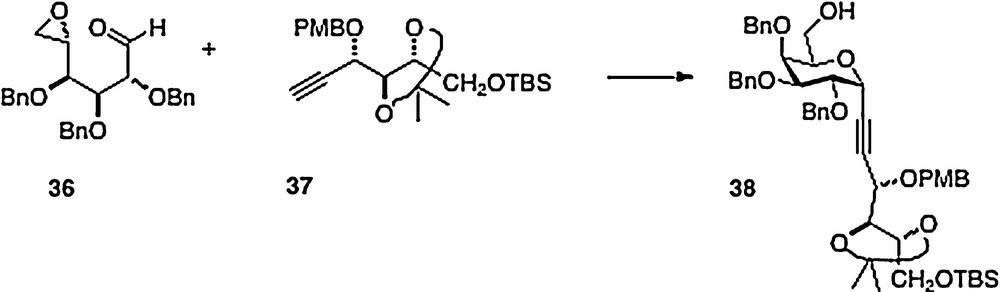
An original and clever synthesis in this series is described by Haudrechy [15]. Thus, the acyclic epoxide/aldehyde 36 derived from galactose is smoothly condensed with arabinose-derived alkyne 37 to afford alcohol analog 38 (Scheme 7). This material provides, in a few straightforward steps, ester analogs of α–C–galcer in place of the typical amide function. If the initial condensation were to be done with a ribose-derived alkyne, then presumably the alcohol product could be converted to an amine with inversion of configuration to obtain entry into the “parent” galcer series.
There is an analogous regioisomeric construct to the C-glycoside design/hypothesis which presumes that the pyranose ring O can be replaced by CH2 to prepare cyclitol analogs of glycosides. This concept has been developed by Mori for galactosyl ceramides [16]. His research has led to active materials, but will not be reviewed here.
2 Immunostimulant effects of α–C–galcer
Our C-analogs have been studied in the mouse as a prevention agent for malaria, asthma, arthritis, and ocular autoimmunity, as a vaccine adjuvant in tuberculosis and influenza, and with cancer in mice, either as a single agent for prevention or as an adjuvant in monoclonal antibody therapy. In almost every case, O-galcer has been used as a positive control. The controls verifying the necessity of NKT cells and APC cells with their proper receptors, and the ability to produce the requisite cytokines were carried out in all the studies described below. In several reports, the C-galcer analogs have displayed activity significantly superior to O-galcer, even orders of magnitude better. In other cases superiority of C-galcer was modest, or minimal difference was observed between C- and O-galcer, within experimental error. It is fair to say that in no case did O-galcer appear to be a better immunostimulant in directly compared assays. There are reports of O-galcer in other disease models where C-galcer has yet to be tested. In general, immunologists have classified responses to glycolipid materials as being of two types, TH1 for infectious diseases and cancer, TH2 in cases of autoimmune disease. The TH1 response is expected to be strong when induced by IFNγ and IL-12 while the TH2 effect is signaled for by IL-4. In those cases where a TH2 response, induced by IL-4 is determining, one would expect little or no effect to be stimulated by C-galcer or any of its analogs which uniformly do not promote IL-4 production. In the specific case of OCH, the O-analog with a truncated phytosphingosine initially was reported to stimulate secretion of IL-4 but no other cytokines. One would thus expect its effect on experimental autoimmune encephalomyelitis to be superior to C-galcer which does not demonstrate IL-4 production. However, this prediction needs experimental verification [17].
The malaria studies were carried out by Tsuji et al. and were based on a prevention model [18]. Thus, adult mice were first dosed with either the O- or C-galcer. Then after an interval of 10, 7, 4, or 1 day, the mice were challenged with sporozoites of P. yoeli, the stage of malaria which grows in the liver. After a period of 40–42 hours to permit establishment and growth of the sporozoites, the mice were sacrificed and their livers were assayed for the presence of sporozoites. Table 1 shows the results which confirm a classic C-glycoside behavior; i.e. a longer term effect for a C- compared to O-glycoside. In this example, protection against sporozoites was maintained by the C-galcer for 4 days whereas the O-glycoside offered good protection for only 1 day. Perhaps more interesting, and not predictable, is the dose / response comparison shown in Table 2 where the C-glycoside is active at the nanogram level, 1000-fold more potent than the O-glycoside. This is by far the most profound C-glycoside effect ever reported. It should be noted that C-galcer-protected mice, if not sacrificed, show no signs of blood-stage malaria after several weeks.
Time frame of maintenance of antimalarial activity of galcers.
| Daysa | O-galcer countsb | C-galcer countsb |
| 10 | 1,800,000 | 1,600,000 |
| 7 | 2,000,000 | 1,400,000 |
| 4 | 1,500,000 | < 100,000 |
| 1 | < 100,000 | < 100,000 |
a “Vaccination” with 1 μg of glycolipid, days prior to challenge with P. yoeli sporozoites.
b Counts of RNA of sporozoites of P. yoeli.
Dose/response of antimalarial activity of galcers.
| Dosea | O-galcer countsb | C-galcer countsb |
| 1000 | 100,000 | < 1000 |
| 100 | 300,000 | < 1000 |
| 10 | 500,000 | < 1000 |
| 1 | 750,000 | 20,000 |
a “Vaccination” with nanograms of glycolipid, 1 day prior to challenge with P. yoeli sporozoites.
b Counts of RNA of sporozoites of P. yoeli.
The asthma studies in mice were conducted by Umetsu et al. [19]. The asthma model in mice requires an initial sensitization of the 6-week-old mouse with OVA followed about 2 weeks later with a second sensitization by OVA and then a challenge with methacholine. At this point asthma-like symptoms occur. In the Umetsu protocol, the O-and C-galcers are administered to 2-week-old suckling mouse pups. The C-galcer 39 was used in this assay. Then the sensitization/challenge protocol is followed and the 8-week-old adult mice are observed for asthma symptoms. The results show that the C-galcer is protective to a greater extent than the O-galcer parent and a novel O-glucoside 40 originally isolated from Helicobacter pylori.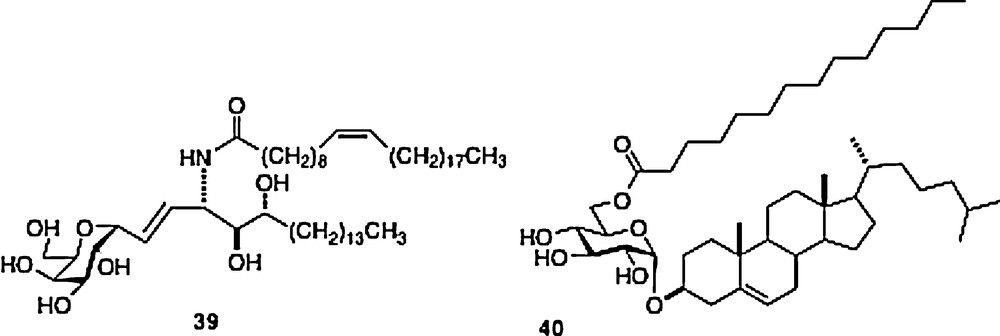
This last material is protective, but requires a 10-fold higher dose than C-galcer. In parallel experiments, Umetsu et al. showed that infection of the suckling mouse pups with H3N1 influenza also protected the mouse from adult asthma subsequent to antigen challenge. In a very interesting extension of this protection scheme, an adoptive transfer experiment was undertaken. Thus, NKT cells were isolated from 8-week old mice that had been protected at their 2-week suckling stage by C-galcer 39 or O-gal 40. These cells were injected into naive 8-week old mice which were then challenged with allergen. The protective effect was transferred with the cells. These data suggest that the effects of C-galcer 39 and 40 are more profound than their stimulation of cytokine secretion upon formation of the ternary complex described above. There seems to be a “memory effect” revealed in these experiments.
The effects of C-galcer in the mouse arthritis model were reported by Elewaut et al. [20]. The experiment uses a collagen injection to initiate arthritis-like symptoms in the mouse which is then treated with either O- or C-galcer 5 days postchallenge. The animal is then observed for development of disease. After displaying a full-blown disease, the animal is euthanized and an autopsy is performed. Both O- and C-galcer were shown to ameliorate the severity of the arthritis, with no major difference between the two ligands in their potency. The authors postulate that the mechanisms of amelioration are different because the levels of cytokines induced by the glycolipids are different. If the galcer treatment were delayed till day 20 postchallenge, the galcers were no longer effective.
Experiments with a mouse model of human autoimmune uveitis have been described by Caspi et al. [21]. Mice are treated with an interphotoreceptor retinoid-binding protein (IRBP) with or without added glycolipid. After 19 days, the eye fundus is examined for uveitis. With this protocol, 18/18 control mice develop eye disease whereas 12/18 mice contract eye disease when protected with O-galcer. C-galcer limits disease to 9/18 animals and the overall severity of disease is reduced.
The TB protection experiments were reported by Porcelli et al. [22]. The protocol uses BCG as a vaccine with or without incorporated glycolipid. The mouse is first injected with the candidate vaccine followed by challenge with an aerosol of TB 2 months after vaccination. Then the mouse is sacrificed either 3 weeks or 6 weeks postchallenge at which point the lungs and spleen are examined for TB. Compared to control, all three drug formulations reduced the amount of TB. Both O- and C-galcer – enhanced BCG were more potent than BCG alone in reducing quantifiable disease. However, in every case, the vaccine combination efficacy diminished in the interval between 3 and 6 weeks postchallenge, implying that protection was only temporary. Interestingly, the C-galcer revealed the greatest potency in the spleen at the 3-week interval, more than twice the protective effect of BCG or BCG-O-galcer. At the 6-week spleen assay, the C-galcer remained the most potent. In the lungs, C-galcer was more potent than the others at the 3-week assay and equipotent with O-galcer at the 6-week assay.
The influenza vaccination tests were performed by Kopecky-Bromberg et al. [23]. The vaccine was a live, attenuated influenza virus which by itself at a dose of 104 PFU (plaque-forming units) protects mice from influenza challenge 3 weeks post-vaccination. However, at lower doses, the vaccine itself is not effective. When the experiment is performed with vaccine supplemented with C-galcer, a useful protection is observed. A series of dose-response experiments revealed the following interesting results. With a vaccine dose of 100 PFU complemented with 1 μg of glycolipid, four of five mice survive the challenge with wild-type influenza. A complement of 2 μg of C-galcer only protected two mice while 4 μg did not protect the mice, which all died. However, with larger doses of vaccine, 103 and 104 PFU, the high-dose of C-galcer did not lead to mouse death. In further dose-response experiments where the vaccine component is lowered to 25 PFU, a complementary C-galcer supplement of 0.33 μg protected four of five mice from influenza challenge, 1 μg protected three mice and 0.11 μg or 3 μg protected only one mouse. These data seem to suggest that C-galcer has a positive low-dose effect on flu vaccine and a deleterious high-dose effect. It is worthy of note that in the best combination of vaccine (25 PFU) and C-galcer (0.33 μg), the amount of vaccine required for mouse protection from influenza is reduced by a factor of 40 (25 PFU vs. 1000 PFU). By comparison, O-galcer has been tested similarly with a flu vaccine, but with a protocol that uses three successive weekly doses of vaccine/O-galcer prior to challenge. The doses range from 0.125 μg to 2 μg of O-galcer. The results of flu protection are about the same as for the C-galcer. Interestingly, the 2 μg dose is a bit less effective than the 1 μg dose, suggesting the same kind of response curve with both O- and C-galcers [24].
Two different varieties of experiments with O- and C-galcer were done with cancer models. The Tsuji lab described a prevention experiment [9]; mice are treated with either C-galcer or O-galcer and then challenged with melanoma cells. In this model the melanoma grows in the lungs of the animal. After 3 weeks, the mice are sacrificed and the lungs are examined visually for individual melanoma black spots. A dose of 100 ng of C-galcer kept the lungs free of melanoma, whereas a 10 ng dose was not totally preventative. A 1 μg dose of O-galcer was about as effective as the 10 ng dose of C-galcer. These results were independently confirmed by the Steinman lab [25] (Fig. 2). Neither glycolipid is useful against pre-established melanoma disease.
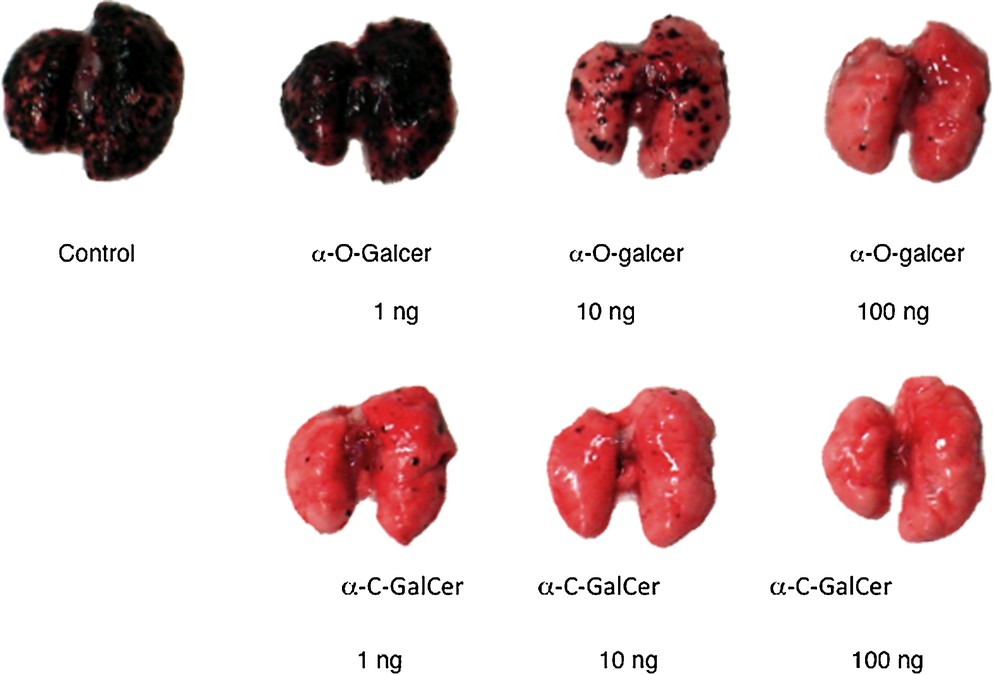
Lungs of Balb mice 3 weeks after pre-challenge treatment with glycolipid followed by challenge with melanoma cells (reprinted from [9] with permission).
A cooperative and curative effect of C-galcer was described by Smyth et al. [25a]. This group had shown that a triple cocktail of monoclonal antibodies (anti-DR5/anti-CD40/anti-4-1BB) could eliminate established kidney or breast tumors in all or almost all mice in a test group, whereas omission of anti-CD40, the most toxic of the three, rendered the cocktail ineffective. In the event of substitution of the missing antibody by C-galcer at the 25 ng level, the elimination of tumor was again observed with the rescue of four of five mice. By comparison, O-galcer at the same dose was not effective, but at an increased dose of 50 ng, three of five mice responded. In a related experiment, the Smyth and Johnstone labs [25b] showed that the combination of mAbs anti-CD40 and anti-CD137 enhanced the tumor regression behavior of the histone deacylase inhibitors vorinostat and panobinostat. Again, replacement of anti-CD40 with C-galcer produced tumor regression (s.c. colon model). It is interesting to compare dosages. Panobinostat: 10 mg/kg everyday for 5 days, a 2-day break, then repeat 5 days of dose. Antibodies: anti-CD40 25 μg and anti-CD137 100 μg every 4 days for a total of four doses. C-galcer: 500 ng as per antibody schedule.
3 Interpretive discussion
Prior to the completion of our synthesis of C-galcer and its first immunology assays, our expectations for its activity were based on the literature of C-glycoside analogs of bioactive O-glycosides. The general consensus among practitioners was that the significant activity difference between the C-glycoside and its O-glycoside “parent” compound would be that the C-analog, being stable to cleavage, should have a similar but prolonged activity compared to the O-glycoside [26]. Thus, in our own earlier work in the 1990s and in a seminal paper by Bertozzi and Bednarski which are typical [27], the C-analogs had very similar activity to their O-glycoside lead compounds. There has also been a literature discussion about the degree of conformational similarity in the aglycone region of the C-compared to O [28]. Clearly, the C-analog would have one fewer H-bond acceptor and would lack both endo- and exo-anomeric effects. But there had been no expectation for a very large difference in biological activity between the analog and the parent. Therefore, the results described above for C-galcer were a pleasant surprise.
The first-order explanation for the difference in activity between O- and C-analogs is that the C-glycoside has a significant divergence from the “parent” O- in cytokine profile. In a series of in vivo assays of all the C-analogs, their IFNγ levels were found to be higher than that for O-galcer, about 2–4 fold at the best, along with a longer period of secretion [29] (Fig. 3).
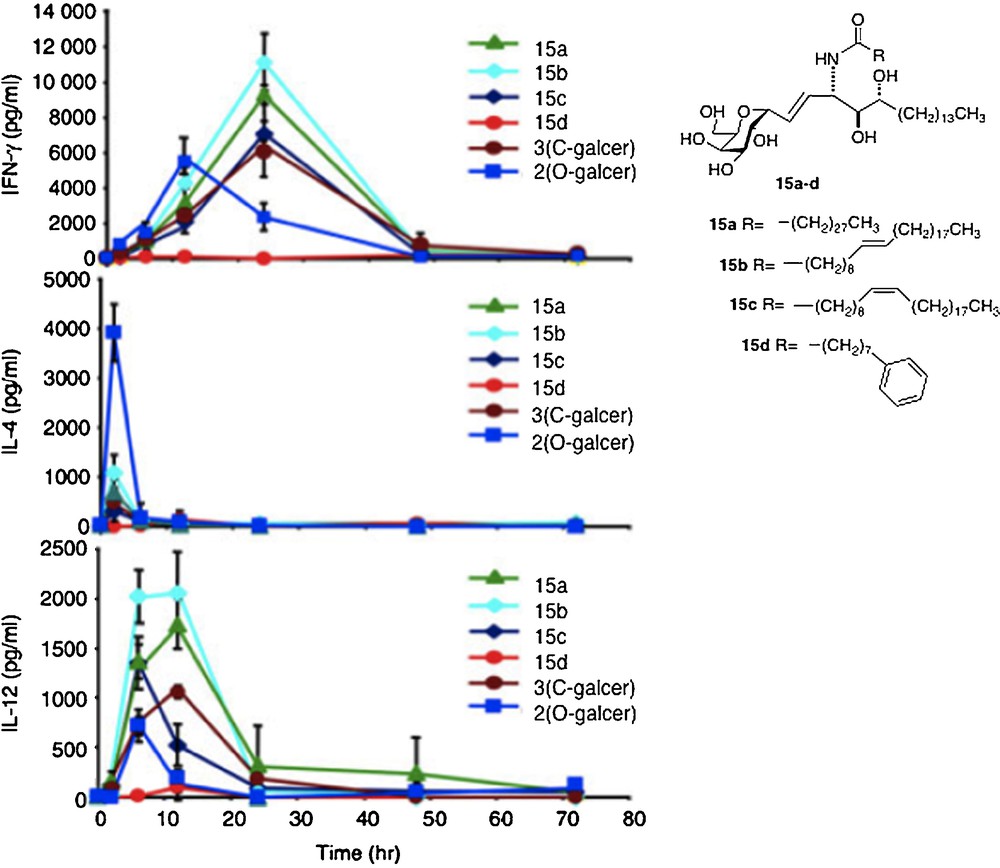
Cytokine levels measured in sera of Balb mice after injection of glycolipid (reprinted from [30] with permission).
The IL-4 levels of the C-glycosides were barely detectable in most cases and far lower than O-galcer. For C-galcer, IL-12 levels were 3–5 fold higher, again with a longer secretion period than the O. Several labs attributed the improved performance of C-galcer to the low levels of IL-4. Thus, it is postulated that IFNγ signals for a THI response of the immune system whereas IL-4 signals for a TH2 response and that the two signals are antagonistic. Since O-galcer causes the production of both cytokines, it would be expected to be less effective whereas the C-galcer, producing only IFNγ, presents a pure TH1 signal, hence more effective. This hypothesis was tested in the malaria system by Tsuji [18b]. Thus, a mutant strain of mouse that is unable to produce IL-4 was subjected to the malaria assay with O-galcer and C-galcer. In the event, the C-galcer remains the better inhibitor of sporozoite growth. In contrast, when a mutant mouse which is unable to produce IL-12 is used in the malaria assay, then O-galcer and C-galcer are equally poor inhibitors of sporozoites. Thus, at least in the malaria example, the hypothesis of IL-4 / IFNγ antagonism is not explanatory. The cancer prevention experiment was also carried out with the IL-12 negative mutant mouse. Again, the C-galcer was no longer a potent drug. Hence, the conclusion one is led to, based on these limited data, is that the signal by C-galcer for increased IL-12 secretion is the basis for its improved performance.
For the structural explanation as to why C-galcers present a different cytokine pattern, it is not clear that its presumed resistance to cleavage could be a cause. Another postulate is that the difference in binding constant, either of the ligand to the CD1d receptor or of the active ternary complex can be an explanation. In fact, there seems to be a weak correlation in the difference of activity between O- and C-galcer in that the former ligand seems to be the tighter binding ligand in the ternary complex, based on in vitro data, but only signals for greater production of IL-4 but not for greater IL-12 or IFNγ [30]. Interestingly, using displacement experiments in vivo, both Steinman and Kronenberg groups showed that the O-galcer was not a stronger binder than C-galcer [25,31]. There is a correlation in the different binding constants of human and mouse CD1d/ligand/NKT complexes and cytokine levels for the C-galcer ligands 41 and 42. Ligand 41 has a stronger binding constant in the mouse in vitro assay and produces the higher cytokine secretion, while ligand 42 with its stronger binding in the human cell in vitro assay signals for the higher cytokine secretion. In the C-gal series, such differences may explain the weak effects on human NKT cells of the parent C-galcer [32].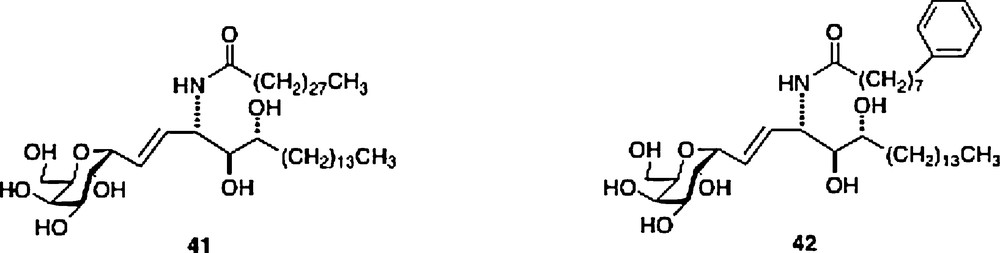
There is a small structural difference between ternary complexes of O- and C-galcer as seen in their x-ray structures [33] (Fig. 4). The visible difference is in the conformations in the 3′,4′ hydroxyl region in their phytosphingosine side-chains. This difference seems to then be propagated in subtly different hydrogen bond networks in the NKT receptor. The O-galcer may have a weak H-bond to the anomeric O-link, which cannot exist in the C-analog [34]. How these small structural differences then lead to different cytokine levels is not explainable at this time.
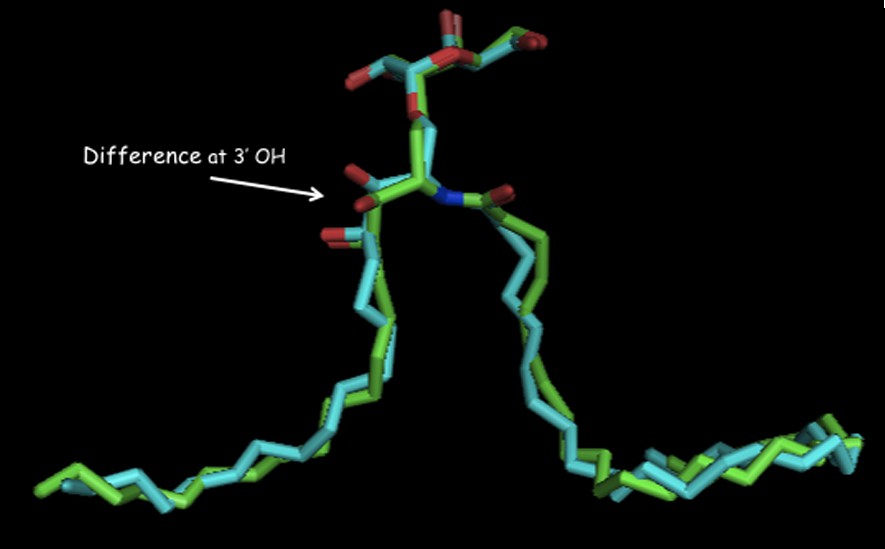
Overlay of ligands bound in their ternary complexes: O-galcer: blue, C-galcer: green (shown with permission of D. Zajonc).
In conclusion, the C-galcer family has produced some interesting synthetic chemistry and some surprising immunology effects. There are two goals for future development of the C-galcer family. First, for a C-galcer to eventually become a drug, there needs to be a demonstration of good immunology activity by a C-galcer in mammals other than mice. It should be noted that O-galcer has been the subject of a small number of human clinical trials. Although little positive effect was observed, there was the significant outcome that no toxic reactions to the drug were detected [35]. Second, with a crystal structure of the ternary complex of C-galcer now available, there should be a design/synthesis effort to alter binding and the resulting hydrogen bonding network of the NKT receptor to try to gain a more thorough understanding of why C-galcer works so well, and thus to guide the preparation of even more potent analogs.
Acknowledgement
The synthetic chemistry research at Hunter College has been supported by a grant to RWF from NIH (GM 60271), by a subcontract to RWF from the NIH Tetramer Core Facility at Emory (NO1-AI-25456), by contract funding to RWF from Cytheris, S.A and by an infrastructure support grant from the NIH to the Hunter College RCMI program (RR 03037). The critically important immunology program was begun at the lab of Moriya Tsuji (NYU, ADARC) and then expanded to the Steinman lab (Rockefeller), the Kronenberg lab and the Zajonc lab (La Jolla), the Smyth, the Rossjohn, the Godfrey and the Johnstone labs (all in Melbourne), the Umetsu lab (Harvard), the Caspi lab (NIH), the Palese lab (Mt. Sinai), the Elewaut lab (Ghent), the Porcelli lab (Albert Einstein). To all these labs and funding agencies, the author is sincerely grateful.