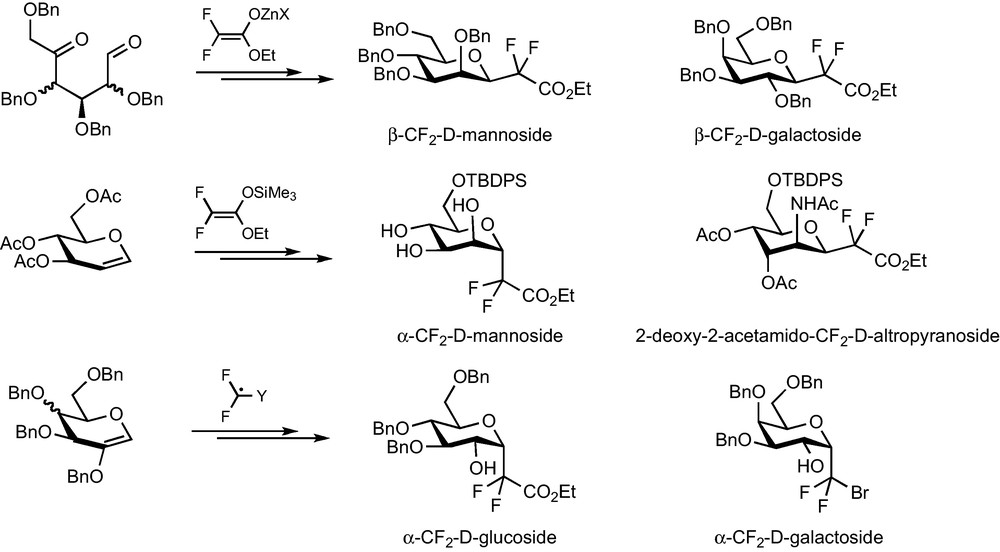
1 Introduction
The incorporation of fluorine atoms onto biomolecules or synthetic biologically active molecules has become an intense research field in the two or three past decades, especially for drug development purposes [1]. This widespread interest stems from the unique properties of the fluorine atom and of the C–F bond [1,2]. With a van der Waals radius of 1.47 Å, the fluorine atom is small (intermediate size between the hydrogen and oxygen atoms) but strongly electronegative (electronegativity of 4.0 on the Pauling scale) and exhibits a high electron affinity (Eea = + 79.5 kcal.mol−1). This results in a short (1.35 Å) and highly energetic C–F bond (dissociation energy is around 105.4 kcal.mol−1, the strongest bond of organic chemistry) with a marked ionic character. As a consequence, the introduction of a fluorine atom into a biologically active molecule often allows one to induce alterations of crucial biological properties within limited structural modifications [2]. An increase in the lipophilicity, the modulation of the acidity/basicity or the prevention of metabolic degradations due to the presence of a fluorine atom in the appropriate position can indeed often induce an improvement of the pharmacodynamic or pharmacokinetic properties of the molecule [3].
Due to the implication in many biological events of their natural counterparts, fluorinated carbohydrate derivatives have thus received careful attention whatever the nature of the fluorinated substitution or its position on the sugar backbone [4]. A fluorine atom or a fluoroalkyl group can for example be introduced as a surrogate to a –OH or –CH(OH)– group in order to probe the interaction between the glycosidic compound and its biological target. Fluorination at C-2 has led to the well known radiopharmaceutical 2-fluoro-2-deoxyglucose (18F-FDG) and is very efficient to slow down the metabolic degradation of glycosidic compounds. As far as metabolic stabilization is concerned, a more drastic modification to the glycosidic structure is the replacement of the osidic oxygen by a carbon link to provide the so-called C-glycosides [5]. For that matter, the use of a fluoroalkyl group (CF2 or CFH) has often been considered to provide surrogates with better mimicking abilities than classical C-glycosides, thanks to the above-mentioned electronic properties of fluorine. To try and challenge this hypothesis, an important synthetic work devoted to the preparation of such compounds has been reported [6]. Among these different approaches, our team has a long-standing interest in providing methodologies which allowed their synthesis for many carbohydrate series (glucose, mannose and galactose) and for any pseudo-anomeric center configuration (α or β). Due to the striking results obtained by the Franck group with C-glycosidic analogues of α-galactosylceramides, the preparation of their fluorinated counterparts appeared to us as a nice opportunity to compare these two classes of glycomimetics and as an exciting application for fluorinated surrogates. We wish to present herein an overview of our efforts devoted to the synthesis of CF2-glycosides, which has recently led to the disclosure of an efficient methodology for the synthesis of analogues of α-galactosides and α-galactoconjugates.
2 Our strategy
Our work on the development of synthetic methodologies for the preparation of CF2-glycosides relies on several guidelines. First, any methodology should be potentially applicable to the synthesis of complex glycoconjugate analogues and therefore should lead to a CF2-glycosidic intermediate featuring an appropriate functional group in pseudo-anomeric position. The second prerequisite was that we should stick to fluorinated synthon-based methods and step aside from electrophilic or nucleophilic difluorination methods which are often unproductive on hindered and functionalized substrates such as carbohydrate derivatives. Finally, de novo syntheses of the sugar backbone were also ignored as they would require a higher number of steps to reach these intermediates. Several methodologies based on the introduction of a functionalized, easily accessible fluorinated synthons (BrCF2CO2Et, CF2Br2…) to a glycosidic substrate were therefore developed and reported (Scheme 1).

3 Reformatsky-type additions
The availability of ethyl bromodifluoroacetate and the efficiency of the related Reformatsky reaction prompted us to perform the addition of this zinc reagent to various carbonyl compounds. Acyclic carbohydrate-derived aldehydes required a subsequent ring closure to provide the desired C-glycosidic compounds whereas the addition to glyconolactones directly afforded such compounds.
3.1 Addition to glucose and galactose-derived enals
Our first synthesis was based on the Reformatsky addition of BrCF2CO2Et to enals 1 and 2 prepared in six steps from O-methyl-d-glucose and O-methyl-d-galactose [7]. The reaction is efficient (64% yield on 1, 98% yield on 2) but not diastereoselective, providing both epimers at C-1 of compounds 3 and 4 (Scheme 2) [8]. Modifications of the procedure (low temperature, SmI2-mediated reaction…) did not improve the selectivity, but the two diastereomers could, however, be separated by chromatography. The absence of diastereoselectivity, if disappointing, might at least allow the preparation of α- and β-C-glycosides. Ring closure was thus attempted on each diastereomer. Epoxidation of the terminal double bond of 3a and 3b followed by acid-catalyzed cyclization afforded CF2-glycosides 5, once again with no selectivity for the epoxidation reaction. This 1:1 mixture of C-5 epimers being unproductive, oxymercuration reactions were explored to perform the ring-closure. The reaction was this time diastereoselective and the stereochemical outcome of the reaction appeared to be governed by the C-4 stereogenic center [9]. Indeed, if the Hg(OTFA)2-mediated cyclization of 3a and 3b afforded compounds 6 with a l-idose configuration, the same reaction on 4a and 4b led to the desired d-galactoside precursors 7. An oxidative demercuration reaction on 6 ultimately led to l-idose analogues 8 but could not be performed on 7 due to a lack of time and material.
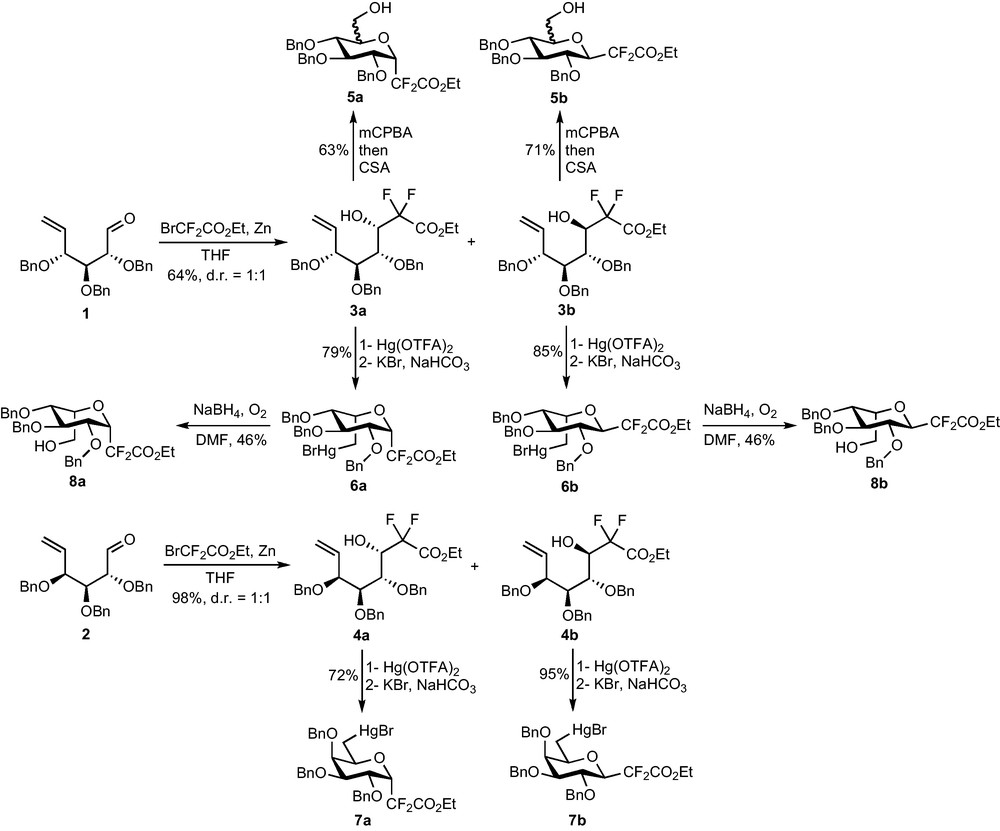
This first access to functionalized CF2-glycosidic compounds was encouraging but the method showed too many drawbacks to be applied to the synthesis of complex glycoconjugate analogues. Indeed, the length of the synthesis (nine steps from the required O-methyl-d-glycoside) added to the numerous problems of stereoselectivity and to the tricky oxidative demercuration step prompted us to find a shorter and more selective access to this intermediate.
3.2 Addition to glyconolactones
A direct addition of the Reformatsky reagent to glyconolactones appeared as a straightforward method to access the fluorinated C-glycosidic backbone. These lactones are indeed prepared in only three steps from the corresponding O-methyl-d-glycosides and their use as substrates for the addition of non-fluorinated Reformatsky reagents was already reported [10,11]. The Zn- or Zn/Cp2TiCl2- or Zn/CeCl3- or SmI2-mediated addition of bromodifluoroacetate was indeed efficient on gluco-, manno- and galactonolactone 9 [12]. The reaction was diastereoselective, providing the compound placing the CF2CO2Et group in pseudo-equatorial position as the major diastereomer, with ratios from 88:12 to 99:1 (Scheme 3). This diastereomer, being probably the most stable one (from a steric and stereoelectronic point of view), it is difficult to determine whether the selectivity is kinetically or thermodynamically controlled. This short and efficient preparation of precursors of CF2-glycosides was, however, undermined by the difficulties which were encountered in the removal of the C-1 hydroxy group. Reductions mediated by silanes and Lewis acids, currently used in standard C-glycoside synthesis [13], were indeed ineffective, despite the numerous conditions which were tested. The strong electron withdrawing effect of the difluoromethylene group probably prevents the formation of the required oxonium intermediate. Classical radical reductions of the corresponding xanthates and oxalates also failed [14]. Though this transformation has been accomplished by others through a SOBr2-mediated bromination and a subsequent reduction with Bu3SnH [15], this transformation can only provide β-CF2-glycosides and a method allowing the preparation of α-CF2-glycosides was still missing.
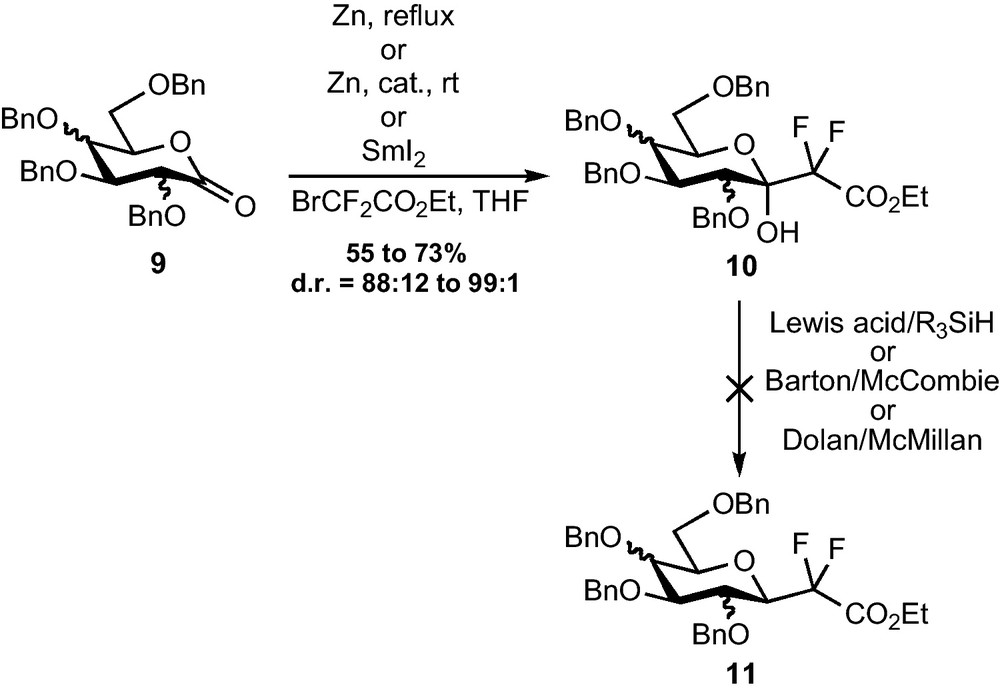
Intermediates of type 10, featuring an additional hydroxy group at C-1, were however converted by our group and others to pseudo-glycopeptides acting as selectin inhibitors [16], ice recrystallization inhibitors [6]l, or involved in other medicinal applications [17].
3.3 Addition to carbohydrate-derived 5-ketoaldehydes
A Reformatsky addition on 5-ketoaldehydes 13, easily prepared in four steps from the corresponding O-methyl-d-glycosides via diols 12, was expected to yield a zinc alkoxide which would spontaneously cyclize to provide 5-hydroxy-CF2-glycosides. A Et3SiH/Lewis acid reduction of the OH group should this time be efficient and allow the preparation of the desired CF2-glycosides. The weak stability of aldehydes 13 forced us to directly engage the crude Swern oxidation product in the Reformatsky reaction. A modified procedure, using a RhI catalyst and Et2Zn for the transmetallation [18], was necessary to obtain compounds 14 and 15 (Scheme 4). The yields were however very disappointing (28% for the mannose series, 32% for the galactose series) and so were the diastereoselectivities (14a:15a = 58:42, 14b:15b = 40:60). Unexpectedly, the reaction was totally ineffective on the corresponding glucose-derived 5-ketoaldehyde.
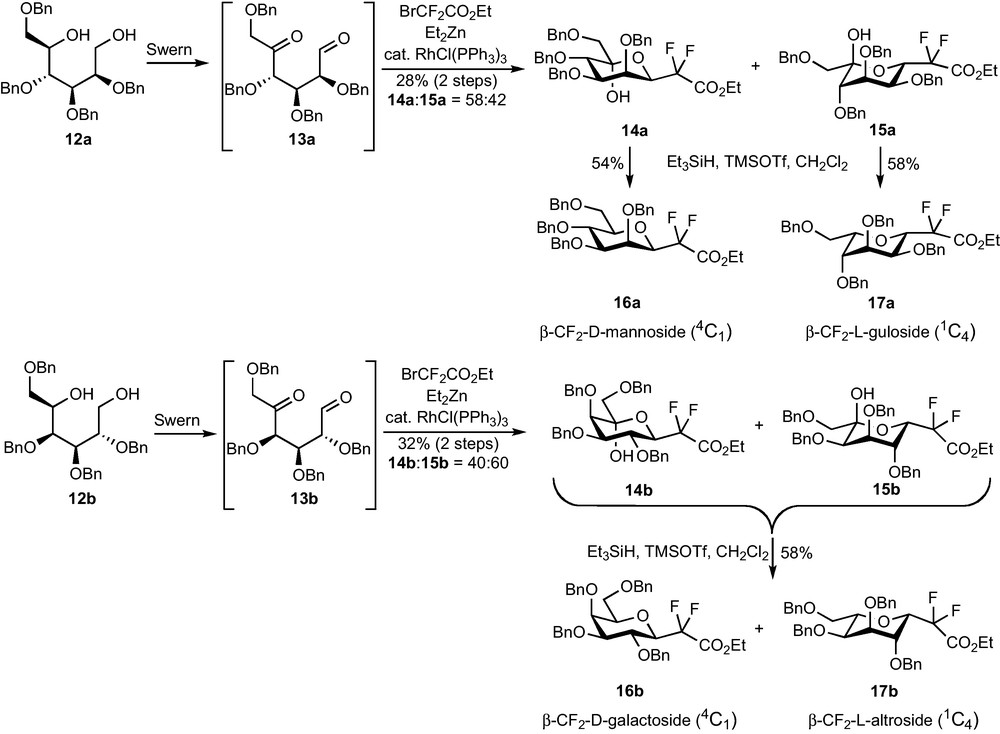
Mannose compounds 14a and 15a could be separated and each diastereomer was subjected to the dehydroxylation conditions, whereas the same reaction was performed on the unseparable mixture of 14b and 15b. Compounds 16a and 17a were obtained in fair yields and as single diastereomers, as well as 16b and 17b which were this time separated by column chromatography. Careful 1H and NOESY NMR studies revealed that, if 16a and 16b were respectively the expected β-CF2-d-mannoside and β-CF2-d-galactoside (4C1 conformation), 17a and 17b were not the corresponding α analogues. An attack on the pro-S face of the aldehyde at the Reformatsky stage probably results in an equilibration towards the most stable conformation and C-5 configuration (Scheme 5), and leads, after reduction, to β-CF2-l-gulopyranoside 17a and β-CF2-l-altropyranoside 17b (1C4 conformation).
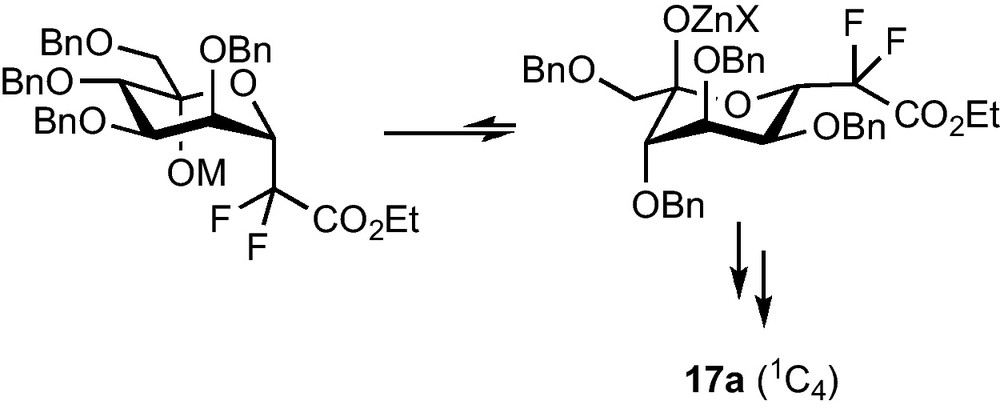
This strategy, despite the low yield obtained in the key step, was applied to the preparation of pseudo-β-mannopeptides as selectin inhibitor [16], but a method that would efficiently allow the preparation of α-CF2-glycosides was, however, still needed.
4 Addition of a difluoroketene silyl acetal
The addition of silyl enol ethers to various glycosyl donors is a known method for the preparation of α-C-glycosides [5,19]. The group of Portella reported in 1996 the elegant in situ generation of a ketone-derived difluoroenoxysilane and its efficient addition to tri-O-acetyl-d-glucal 18 or to 2-deoxyglycosyl donors [6]d. However, and to the best of our knowledge, no functionalization of the double bond resulting from the addition to the glycal has been described by this group. Moreover, such an addition delivered a compound featuring an aryl or alkylketone in the pseudo-anomeric position, which was not suitable for the preparation of glycoconjugate analogues. We therefore decided to explore the addition of bromodifluoroacetate-derived enoxysilane 19 either directly to a glycosyl donor, or to tri-O-acetyl-d-glucal (Scheme 6). The latter would require the investigation of dihydroxylation or aminohydroxylation reactions of 20 in order to obtain CF2-glycosidic intermediates.

Our first task was to identify an appropriate salt-free source of difluoroketene silyl acetal 19 and determine if it was a suitable nucleophile for glycosylation reactions. A modification of the procedure described by Iseki was finally chosen [20]. The Reformatsky reagent was prepared in acetonitrile instead of THF in order to reduce the amount of homocoupling product. Simple extractions with n-pentane eventually provided a salt-free solution of 4 which was used directly for glycosylation reactions, without performing the tedious and poor-yielding distillation of the Iseki procedure. Despite our efforts and in agreement with Portella's report, this nucleophile failed to react with glycosyl donors [6]d. The nature of the protecting group (benzyl, acetates), of the leaving group (acetate, trifluoroacetate, trichloroacetimidate, bromide) and of the Lewis acid (BF3.Et2O, TiCl4, TMSOTf…) had little influence on the outcome of the reaction (Scheme 7). The addition to glycal 19 proved to be much more rewarding since the SN2′ products 20 could be isolated in 72% yield under optimized reaction conditions [21]. The diastereoselectivity was however disappointing (6:4 α:β ratio) and could not be improved despite our efforts.
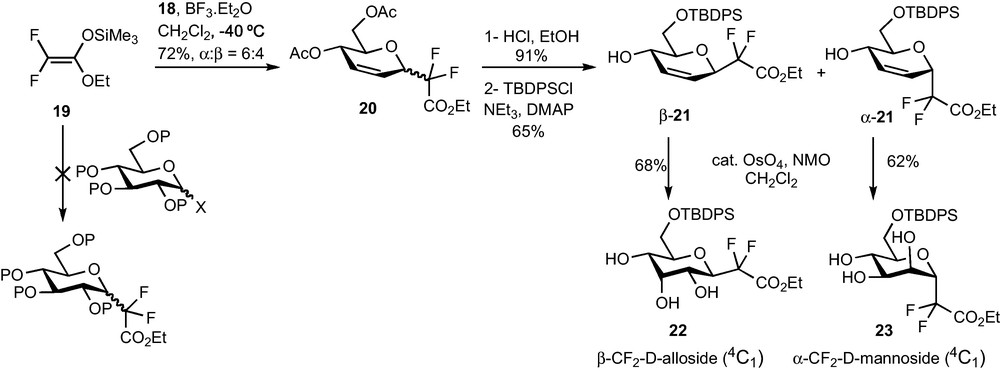
We then decided to submit this unseparable mixture of diastereomers to dihydroxylation or epoxydation reactions, but all attempts were unsuccessful. The poor reactivity of the acetate-protected CF2-glycosylester 20 prompted us to proceed to protecting group modifications. Complete deacetylation and selective protection of the C-6 alcohol with a TBDPS group not only allowed the separation of the two diastereomers α-21 and β-21 but also restored the double bond reactivity (Scheme 7). Osmylation of α-21 and β-21 under Upjohn's conditions afforded CF2-Glycosides 22 and 23 in 68% and 62% yield respectively and only one diastereomer could be detected in each case by 1H and 19F NMR of the crude mixture (Scheme 7). The β-alloside configuration of 22 and the α-mannoside configuration of 23 were determined thanks to HOESY NMR experiments and chemical correlation, and demonstrated that the syn-addition of OsO4 proceeded anti to the CF2 group.
Epoxidation reactions were also investigated and followed the same features as the dihydroxylation reaction. The α/β mixture (6:4) of acetylated compounds 20 was totally unreactive under all the epoxidation conditions which were tested (m-CPBA, V(acac)3/t-BuOOH, MnSalen/m-CPBA, Oxone, Oxone/Trifluoroacetone). On the other hand, deacetylated compounds α-21 and β-21 underwent the desired reaction using methyl(trifluoromethyl)dioxirane (generated in situ from trifluoroacetone and oxone) [22]. Epoxides 6 and 7 were indeed isolated as a single diastereomer for each reaction in 77 and 86% yield respectively (Scheme 8).
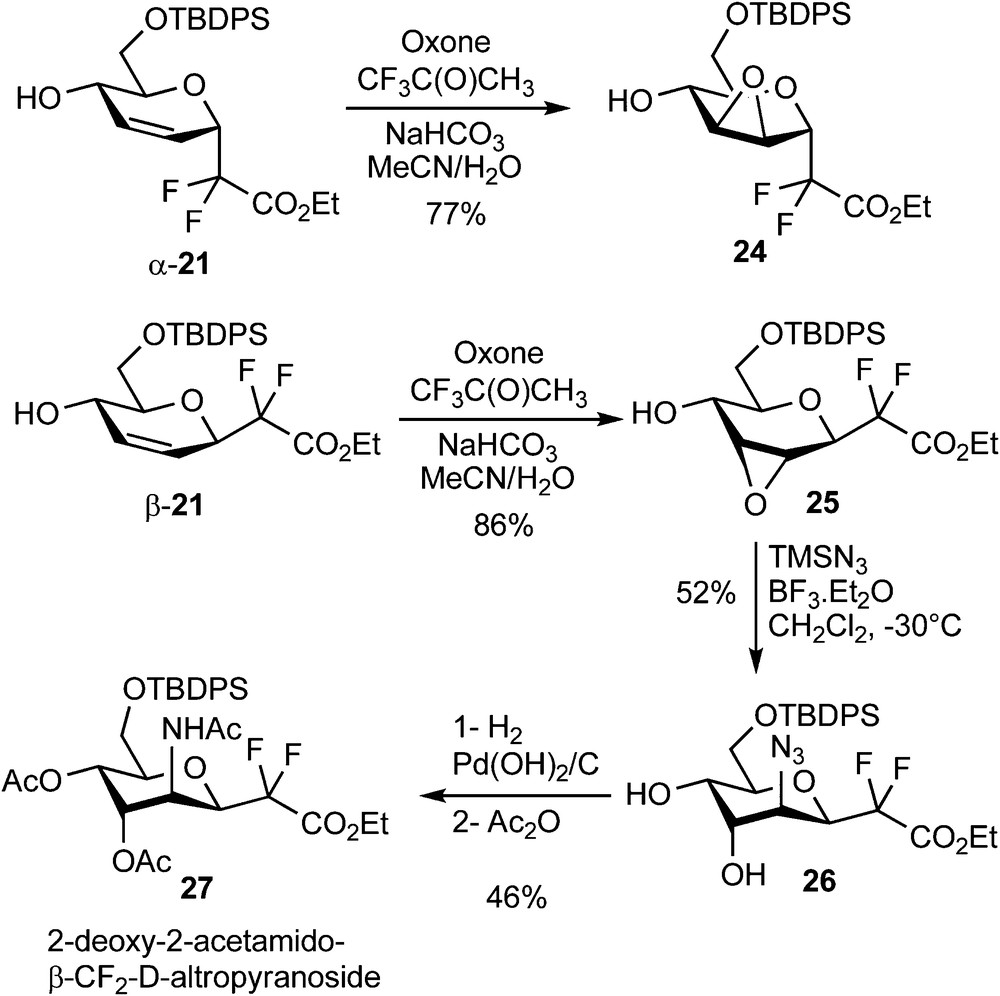
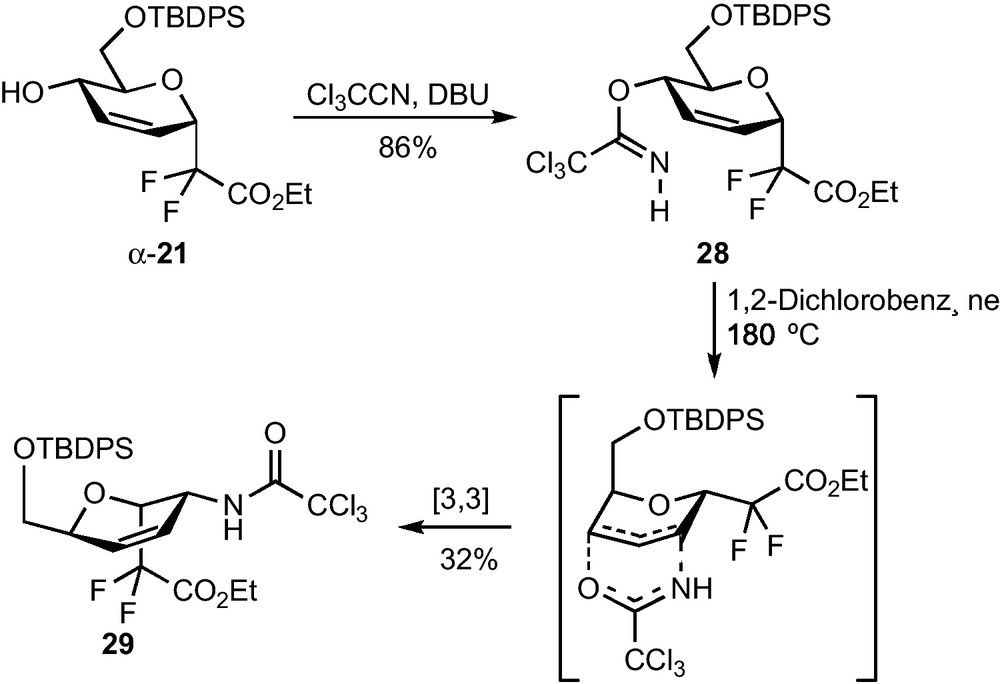
Ring opening reactions using nitrogen nucleophiles were then investigated on compound 25. Among the various nucleophiles (BnNH2, TMSN3, NaN3) and acids (NH4Cl, BF3•Et2O, LiClO4, Yb(OTf)3) which were tested, the best result was obtained using trimethylsilyl azide in the presence of a stoichiometric amount of BF3•Et2O at low temperature. These conditions yielded, as demonstrated by 1H and NOESY NMR experiments, the 2-deoxy-2-azido-β-CF2-altroside 26 in 52% yield and as a single regio- and stereoisomer (Scheme 8) [23]. Hydrogenation and acetylation of 26 yielded 27, which is, to the best of our knowledge, the first reported example of a 2-deoxy-2-acetamido-CF2-d-glycoside. To our dismay, the α epimer 24 was poorly reactive and all our efforts devoted to the ring-opening of this epoxide were unsuccessful. An Overman sigmatropic [3,3]-rearrangement of the trichloroacetimidate 28 derived from α-21 was also investigated as a possible route to 2-acetamido-glycosides [24]. A high temperature was required to promote the sigmatropic rearrangement which provided trichloroacetamide 11 in 32% yield (Scheme 9) [23]. A pseudo-axial arrangement of the trichloroacetimidate moiety in the transition state is indeed necessary for this reaction, which is there only possible by reaching a pseudo-chair conformation of higher energy. This unsatisfactory yield was not optimized since all our attempts to perform a dihydroxylation of the remaining double bond, which would have provided an access to the desired 2-amino-CF2-glycoside, were unsuccessful.
The difluoroenoxysilane addition to a glucal followed by the functionalization of the resulting double bond thus provided an access to α-mannoside and 2-amino-glycoside analogues. Unfortunately, the addition of 19 to tri-O-acetyl-d-galactal, which could have led after transformation of the double bond to interesting galactose derivatives, only met with failure. In order to finally disclose a methodology allowing the synthesis of α-galactoside analogues, an addition of difluoromethyl radicals to modified glycals was investigated.
5 Addition of electrophilic radicals to 2-benzyloxyglycals
The addition of fluoroalkyl radicals to C–C multiple bonds is a widespread method for the preparation of fluorinated molecules [1,25]. The addition of electrophilic radicals onto standard glycals has also been reported and exhibits a well-defined regioselectivity [26]. The addition takes place exclusively at the C-2 carbon (larger coefficient of the HOMO at this position) and is therefore not suitable for CF2–glycoside synthesis. We made the simple reasoning that the introduction of an alkoxy substituent at the C-2 position should direct the addition to the less hindered C-1 carbon (Scheme 10). The work of Miethchen, based on the same assumption, was not reported at the time we begun this study and this hypothesis still had to be challenged [6f].

The radical addition of bromodifluoroacetate to 2-acetoxy-d-glucal 30 or 2-benzyloxy-d-glucal 31 was investigated and a survey of conditions revealed that adducts 32 could be obtained in fair yield when using triethylborane as the radical mediator in a polar solvent (Scheme 11) [27]. Adducts 32 resulted from a fragmentation of the radical obtained after addition to release the carbonyl function and a stabilized tolyl radical [28]. The addition exhibited a modest α selectivity since the two separable diastereomers 32a and 32b were present in 3:1 ratio in the crude mixture (measured by 19F NMR). Interestingly, this selectivity became complete when the reaction was performed on 2-benzyloxy-d-galactal 33. Compound 34 is indeed obtained in 58% yield and as a sole α epimer, which opened a promising route to α-galactoside analogues. Despite a clean and fast addition of dibromodifluoromethane to 33, compound 35 was obtained in a disappointing 41% isolated yield after purification, probably due to the modest stability of this compound [29]. A monofluorinated C-glycoside 36 could also be obtained in 59% yield, thanks to the addition of the bromofluoroacetate-derived xanthate under Zard's conditions [30].
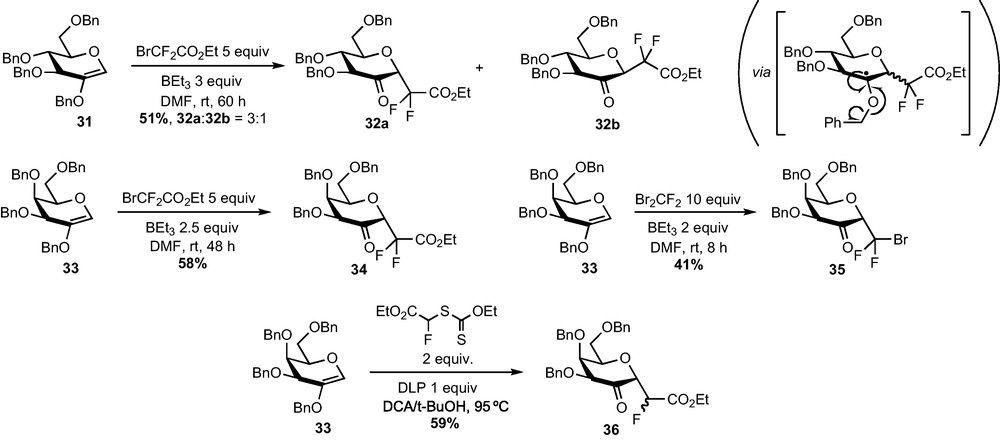
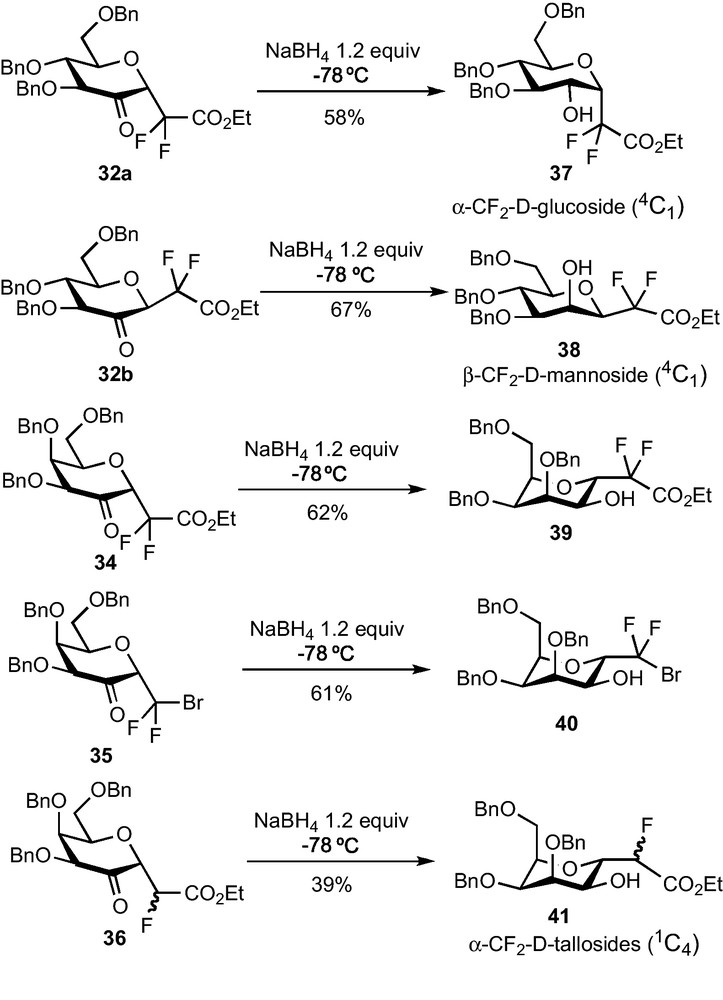
The reduction of 2–ketohexopyranosides 32 and 34–36 to the corresponding C-glycosides was then explored. Simple sodium borohydride-mediated reductions of glucose derivatives 32a and 32b were first examined and afforded respectively the α-CF2-glucoside 37 and the β-CF2-mannoside 38 in a diastereoselective fashion (Scheme 12). In agreement with literature reports, the approach of the hydride proceeds anti to the anomeric substituent, i.e. the CF2 group [31]. Unfortunately, this reaction does not follow the same stereochemical pathway when performed on galactose derivatives 34–36 and α-CF2-tallosides 39–41 were obtained instead of the desired α-CF2-galactosides. Other hydride-mediated reductions were tested (DIBAH, L-Selectride, Et3BHLi…) but led to the same stereoselectivity. Moreover, all our attempts to perform a SN2-type substitution reaction of the OH group at C-2 were unsuccessful. The α-glucoside configuration of 37 was determined thanks to NOESY experiments and confirmed by an X-ray diffraction study. This last analysis revealed that the compound crystallized under a 4C1 conformation, slightly distorted as revealed by the value of some dihedral angles. The α-talloside configuration and the 1C4 conformation of 39–41 was determined thanks to 1H NMR analysis and to the measurement of coupling constants.
As the diastereoselectivity issue we met with kinetically controlled hydride mediated reductions could be tackled under thermodynamic control, Meerwein-Ponndorf-Verley (MPV) reductions were attempted [32]. To our delight, α-CF2-galactosides 42–44 were obtained in good yields and as a single diastereomer in each case (Scheme 13) [33]. A two-step procedure allowed us to significantly improve the global yields, especially in the case of 35. Avoiding the purification of this sensitive compound led to an appreciable 41–53% global yield from galactal 33 (2 steps). The α-galactose configuration of 42–44 was deduced from 1H NMR and NOESY analysis and confirmed by an X-ray diffraction study. These studies also revealed the conformational flexibility of these compounds in solution (some NMR data are compatible with 4C1, 1C4 and 1S3 conformations) and the surprising crystallization of 43 under a 1C4 conformation [6]k.
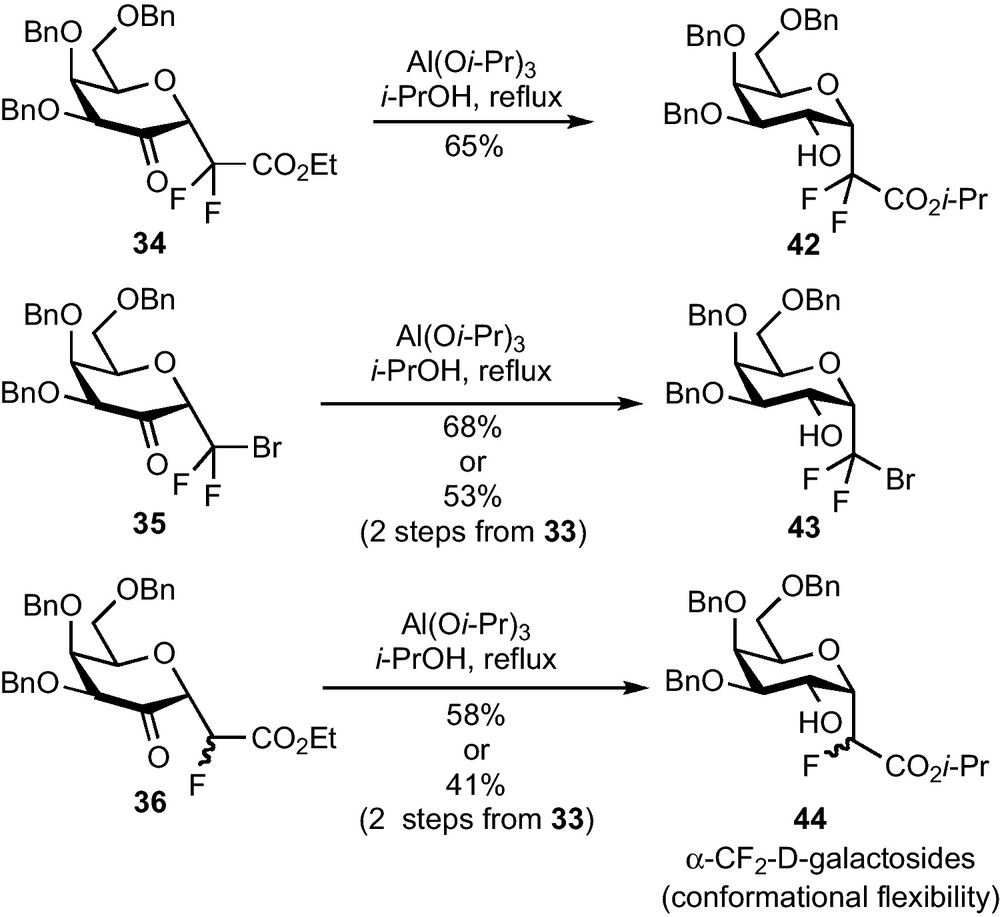
This radical addition/MPV reduction sequence eventually allowed us to prepare functionalized α-CF2-galactosides which could serve as key intermediates for a synthesis of α-galactoconjugate analogues in general, and of α-galactosylceramides in particular. These intermediates were synthesized in only two steps from glycal 33 (five steps overall from galactose pentaacetate) (Glycals 30, 31 and 33 were prepared according to the very efficient method reported by Fairbanks: [34]) and with a complete stereoselectivity for each reaction. An efficient method to perform the introduction of an aglycon in pseudo-anomeric position had yet to be disclosed and the synthetic elaboration of the CF2Y group was investigated.
6 Functionalization of the CF2-glycosidic intermediates
Peptidic couplings and C–C bond-forming reactions from the CF2CO2R or CF2Br moiety, aiming at the preparation of various glycoconjugate analogues, were explored.
6.1 Synthesis of pseudo-glycopeptides
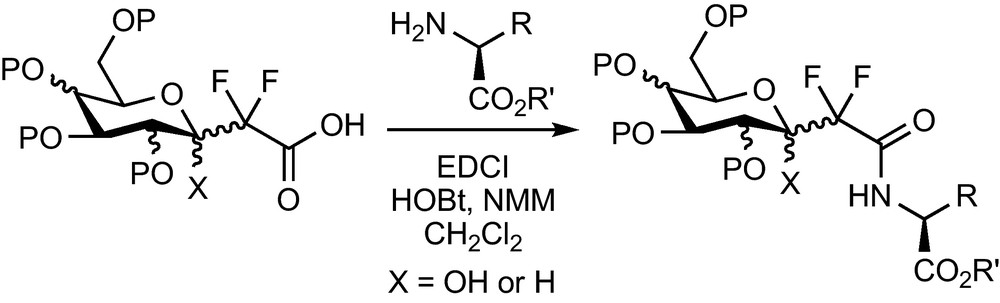
The ester function of the CF2-glycosylacetates prepared previously can be saponified and the corresponding acid engaged in peptidic coupling reactions with a wide range of amines and amino-acids. Many pseudo-glycopeptides, which were designed for various applications, were prepared this way [16,17] (Scheme 14).
6.2 Olefination reactions
Some C–C bond-forming reactions can also be performed starting from these CF2-glycosylacetates. The controlled reduction of the ester function followed by a Horner-Wadsworth-Emmons (HWE) olefination of the resulting hemi-ketal intermediate afforded CF2-glycosides such as 46 with an elongated lateral chain (Scheme 15) [29]. The same sequence is also efficient in the galactose series, using 42 or 44 as the starting material. A hydrogenation of the double bond was performed on 46 to provide 47 in 81% yield.

6.3 Reductive acyl chain transfer from O-2 to C-1’
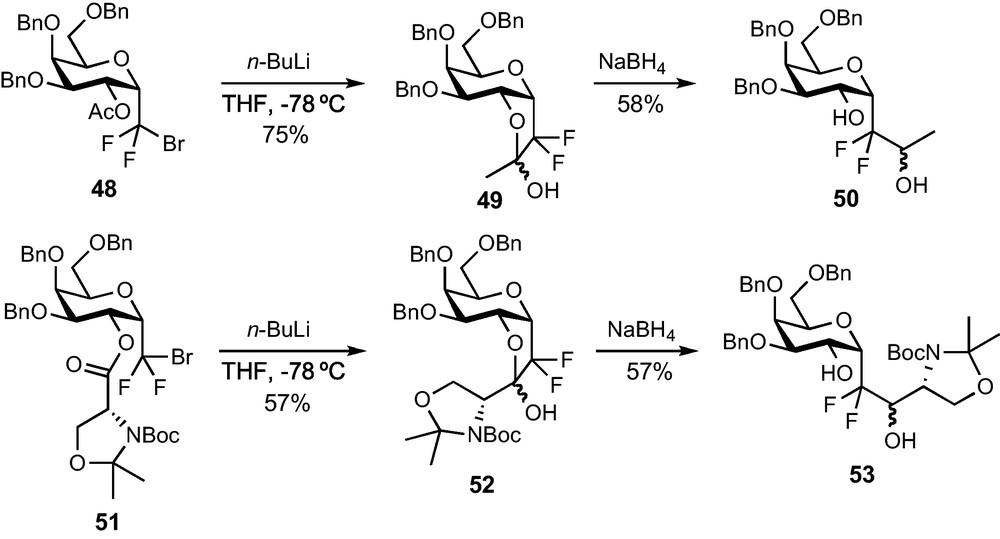
The functionalization of the CF2Br group was also explored since a bromine/lithium exchange followed by an addition on the appropriate electrophile would provide an interesting route to glycoconjugate analogues. If the weak thermal stability of the CF2Li compound made difficult the achievement of an intermolecular addition to aldehydes, an efficient intramolecular process was eventually disclosed (Scheme 16). A Br/Li exchange on acetate 48 indeed resulted in the immediate trapping of the lithium species by the neighbouring acetate to afford the stable hemi-ketal species 49 in 75% yield. A reduction of this intermediate provided alcohol 50 in 68% yield and the global transformation can be looked at as a formal addition of the lithiated anion of 43 to acetaldehyde. Ester 51, featuring a more synthetically useful moiety, was prepared from 43 and from the corresponding d-serine-derived acid (DIPC-mediated coupling, 85% yield) and submitted to the same sequence. Hemi-ketal 52 and the desired functionalized α-C-galactoside 53 were obtained in satisfactory yields (57% for each step) [33]. The sequence is therefore extremely efficient and should allow the introduction in pseudo-anomeric position of various, highly functionalized aglycon moieties through a simple and mild process.
A synthesis of difluorinated α-C-galactosylceramide is currently under investigation in our laboratory using this last methodology. A phytosphingosine-derived acid has been prepared to be introduced on the 43 and to perform the reductive acyl chain transfer to C-1′. This method should allow a rapid introduction of the phytosphingosine chain to the pseudo anomeric position and a straightforward synthesis of α-CF2-galactosylceramide.
7 Conclusion
Several methodologies allowing the synthesis of CF2-glycosides for many carbohydrate series (glucose, mannose and galactose) and for any pseudo-anomeric center configuration (α or β) were thus disclosed (Scheme 17). Throughout this work, we managed to respect our initial guidelines, i.e. the use of fluorinated synthons and of carbohydrate starting materials, preparation of key intermediates featuring an appropriate functional group. All our efforts finally culminated in an efficient preparation of a synthetically useful α-CF2-galactoside intermediate, which was undoubtedly the ultimate goal of this long-standing project. An efficient transformation of these intermediates into α-CF2-galactoconjugates through a reductive acyl chain transfer from O-2 to C-1’ has also been discussed. A route is now opened to prepare the CF2-glycosidic analogues of α-galactosylceramide and compare them to the CH2 analogues developped by the Franck group. The synthesis and biological evaluation of these compounds is currently under investigation in our laboratory and will be reported in due course.