

1 Introduction
Sulfatide is the first sulfated mammalian glycolipid isolated from the human brain by Thudichum in 1884, but chemically characterized only in 1962 [1]. It is also known as sulfated galactocerebroside, 3-O-sulfogalactosylceramide, SGalCer, SGC, or SM4 s. Sulfatide is particularly abundant in the nervous system [2], where in the myelin sheath, it makes up 4–6% of the myelin lipids, but it is also ubiquitously present in various other tissues, such as kidney, pancreas, the gastrointestinal tract and the respiratory tract [3–6].
1.1 Sulfatide structure
Sulfatide is a family of sulfated galactosylceramides, in which a residue of galactose is sulfated at position 3 and is connected through a β-glycosidic bond to the primary hydroxyl group of a N-acylated d-erythro-sphingosine base, typically with a C-18 backbone (Fig. 1). Sulfatide exhibits variation of its structure in the composition of the fatty acid residue that acylates the amino group of sphingosine. The alkyl or alkenyl residues are of different lengths and they can be also hydroxylated. This structural variability shows a cell type-specific pattern, which at least in part can be explained by the different expression of the enzymes involved in sulfatide biosynthesis. The nervonic acid (C24:1)-containing sulfatide is the most abundant in myelin, while significantly elevated levels of stearic acid (C18:0)-containing sulfatide are found in the cortical grey matter [5]. Mammalian kidney sulfatides are instead particularly rich in C22:0 acid, which is found in quantities more than 10 times higher than those in the brain. On the other hand, mass spectroscopic analyses of pancreatic sulfatide revealed a high proportion of short chain C-16 fatty acid and no hydroxylated form [6].
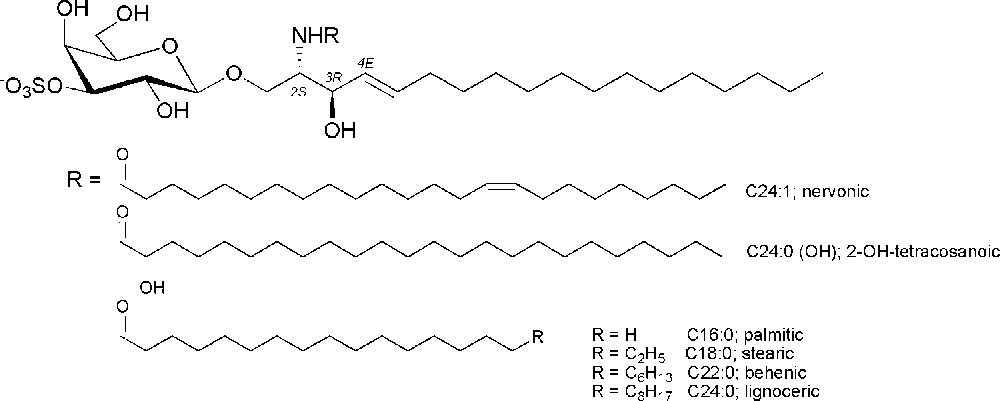
Structures of the glycolipid sulfatide and of the most abundant mammalian fatty acids.
1.2 Biochemical pathways
The synthesis of sulfatide takes place in the endoplasmic reticulum (ER) and the Golgi apparatus. It starts in the ER with the addition of galactose from UDP-galactose to ceramide catalyzed by the UDP-galactose:ceramide galactosyltrasferase (CGT; EC 2.4.1.45) to give galactosylceramides (GalCer). GalCer is then transported to the Golgi apparatus where sulfatide is synthesized by regioselective sulfation catalyzed by cerebroside sulfotrasferase (CST, EC 2.8.2.11). It has been proven that these enzymes have no isozymes. The distribution of sulfatide is tissue-specific and its expression is mainly regulated by the tissue-specific expression of the CGT and CST genes [7].
The degradation of sulfatide occurs mainly in lysosomal compartments within cells where the sulfate group is hydrolyzed by arylsulfatase A (ASA; EC 3.1.6.8). This reaction requires the presence of a sphingolipid activator protein-1 (SAP-1), also called saposin B (SapB), which extracts sulfatide from membranes and makes it accessible to ASA.
1.3 Biological significance
Sulfatide is a biologically multifunctional glycolipid involved in the nervous system, diabetes, virus infection and in the immune system. As the major glycolipid component of the myelin sheath, where it is exclusively found on the extracellular leaflet of the cell membrane, it has a role in the development and maintenance of a compact myelin structure able to exert its functions [8]. The accumulation of sulfatide in the central and peripheral nervous system leads to the human autosomal recessive demyelinating neurodegerative disorder known as metachromatic leukodystrophy (MLD). MLD is a degenerative disease of the central nervous system with essentially motor manifestations and psychosis and is caused by deficiency of the lysosomal enzyme ASA or mutations in the gene encoding for SapB [9]. Furthermore, sulfatide is a major L-selectin ligand in kidney and this interaction plays a central role in monocyte infiltration into the kidney interstitium and hence in the induction of inflammatory response [10]. On another side, sulfatide is also expressed by pancreatic β-cells where it is localized in the same cellular compartment as insulin and is probably involved in processing, maintenance and secretion of insulin [6].
Sulfatide is also involved in human immunodeficiency virus (HIV-1) infection [11]. The entry of HIV-1 into the cell is a complex process mediated by the viral envelop glycoprotein gp120 which interacts mainly with the CD4 molecules at the surface of the cell. Nevertheless, gp120 is also able to bind other receptors including sulfatide. In particular, sulfatide acts as a secondary attachment site during infection, as an alternative binding receptor in the infection of CD4-negative cells of neural and intestinal tissues and participates actively in transmembrane signaling.
Studies on the activation of the immune system mediated by CD1 proteins (CD1s) showed that sulfatide is an antigen of CD1s. CD1 proteins are a family of antigen presenting molecules that after lipid and glycolipid recognition are able to activate the immune cascade in a way different and complementary to the one mediated by the proteins of the major histocompatibility complex (MHC). Sulfatide has been described as a promiscuous ligand of group I CD1 molecules that binds different CD1 isoforms stimulating specific T-cells [12].
Significant changes in sulfatide levels have also been observed in Alzheimer's as well as other diseases, suggesting a role in the pathogenesis of these disorders [13].
This stimulating biological background reveals the need for scientists to have access to quantities of sulfatides necessary for experimental studies. Most biological work has been made with sulfatides from natural sources, e.g., isolated from pig brain [14] or bovine brain [15], or semisynthetic sulfatides, some of them commercially available. This material has been used directly or after simple synthetic transformation, such as the removal of the acyl chain to lyso sulfatide and the re-esterification with the desired fatty acid [6,16] or enzymatic modification of the natural glycolipid [17]. Nevertheless, the material from natural origin can be sometimes contaminated with some isoforms and this has led to the development of synthetic strategies to obtain sulfatide by chemical synthesis in high purity and precise lot consistency.
The aim of this account is to show the synthetic work, correlated to the biological background, that has been described for the preparation of sulfated galactocerebrosides, with particular focus on the syntheses of sulfatide and of some significant related compounds modified on the galactose moiety.
2 Synthesis of 3-O-sulfo-β-d-galactocerebroside (sulfatide)
Careful examination of sulfatide structure clearly suggests that the synthesis involves three different aspects: the preparation of the sphingoid acceptor, the glycosylation reaction with a suitable galactosyl donor and the sulfation reaction. The construction of the lipid part and of the galactosylceramide (GalCer) scaffold are indeed the most difficult task during the preparation, as the sulfation reaction is now well established. Much synthetic work, covered by several reviews from the end of the 1990s [18], has been done in this context. Lately, advances in the synthesis and glycosylation of sphingosine and ceramides, as a consequence of the explosion of the studies on the activation of the immune system mediated by CD1 proteins, have been reviewed in more recent excellent accounts [19]. Herein, only the synthesis of β-GalCer finalized to the preparation of sulfated β-GalCer will be described.
2.1 The sulfation reaction
The presence of the sulfate in position 3 is the characterizing element which differentiates sulfatide from cerebroside (β-d-GalCer). Nevertheless, as mentioned above, the introduction of the sulfate is not difficult to accomplish. It can be easily achieved by reacting the target hydroxyl group with sulfur trioxide-pyridine or trimethylamine complex. This approach requires a protecting group strategy that allows during the synthesis the selective deprotection of the hydroxyl group to be sulfated. In the case of sulfatide, a great advantage, in terms of a significant reduction of the synthetic steps, was introduced by S. Flitsch who developed a method for the regioselective sulfation of glycosides [20]. The treatment of a deprotected galactoside with dibutyltin oxide allows the almost exclusive formation of the five membered cyclic dibutylstannylene acetal among the C-3 and C-4 cis hydroxyl groups. In this complex, the nucleophilicity of the equatorial 3-OH group is enhanced towards acylation, alkylation and silylation. In the case of galactocerebroside (Fig. 2), treatment of the dibutylstannylene acetal with the reactive sulfation reagent Me3N·SO3 gives almost exclusively the 3-O-sulfated derivative, being the 3,6-O-disulfate the only trace side-product and with no sulfation of the ceramide allylic hydroxyl group.
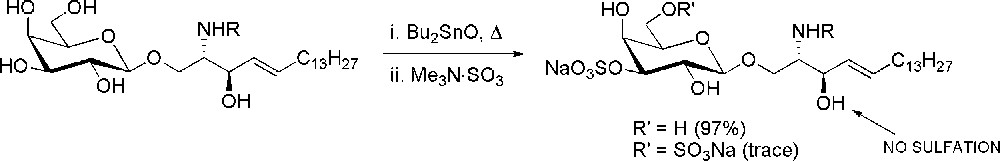
Regioselective sulfation of galactocerebroside via dibutylstannylene acetals.
2.2 The glycosylation reaction
One of the most relevant aspects in glycoconjugate synthesis is the stereocontrol in the glycosylation. In the case of sulfatide, the formation of the 1,2-trans glycosidic bond can be easily controlled and mostly achieved taking advantage of neighboring group assistance by ester protecting group at C-2 of galactose, such as O-acetyl or O-benzoyl. The strategic choice relies mainly in the type of acceptor used in the glycosylation, either azidosphingosine, sphingosine or ceramide and in the selection of the proper galactosyl donor.
The majority of sufatide syntheses have been performed by using 3-O-benzoyl azidosphingosine 1 as the sphingoid acceptor. The “azidosphingosine glycosylation procedure” is generally preferred over glycosylation with ceramide because it provides higher yields. This is mainly due to the non-nucleophilic character of the azido group, which hence does not compete with the primary hydroxyl group towards glycosylation and to the higher nucleophilicity of the hydroxyl group of 1, which is no more involved in intramolecular hydrogen bond formation [18a]. Furthermore, azidosphingosine is an excellent precursor of ceramide, since reduction of the azido function to an amino group is usually easy and the subsequent N-acylation is in general high yielding. The azido group is commonly reduced by Staudinger reaction (usually Ph3P or Bu3P in THF/H2O) or by treating the reaction mixture with hydrogen sulfide in pyridine.
After the seminal synthesis of Schmidt [21], many other preparation of natural (2S, 3R, 4E)-3-O-benzoyl azidosphingosine have been published [22]. These syntheses are all based on the chiral pool approach, which ensures the proper stereochemistry of at least one stereocenter, by starting from naturally occurring chiral substrates (Fig. 3).
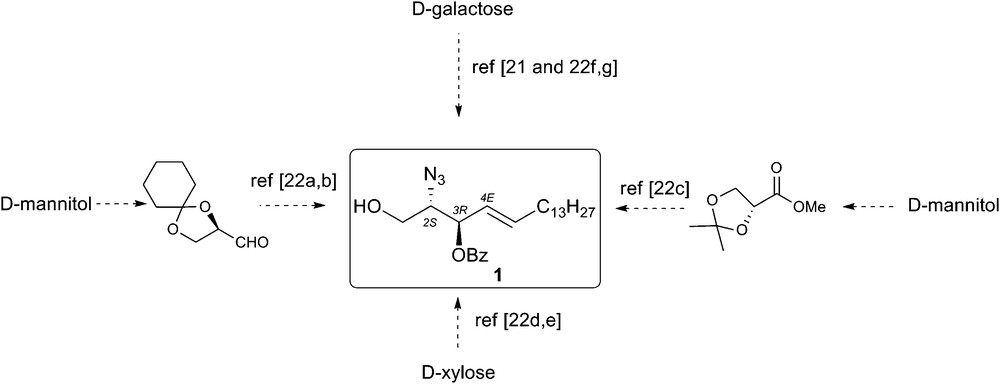
Synthesis of (2S,3R,4E)-2-azido-3-O-benzoyl-octadec-4-ene-1,3-diol.
Scheme 1 gives an overview of the experimental conditions exploited for the glycosidation of 3-O-benzoyl azidosphingosine 1 in the context of sulfatide synthesis. In one of the first examples, tetra-O-acetyl galactosyl bromide 2 was reacted with acceptor 1 under Koenigs-Knorr conditions. The reaction, catalyzed by mercuric cyanide, gave the β-galactoside 3, together with some α-anomer, in 76% and 9.5% yield, respectively [23]. However, glycosyl trichloroacetimidates are the most popular glycosyl donors for glycosidation of azidosphingosine. Thus, the peracetylated galactosyl trichloroacetimidate 4 was reacted with azidosphingosine 1 using BF3·Et2O as the promoter to give the β-glycosylated product 3 in 56%. Yields were slightly increased to 62% when the reaction was promoted with TMSOTf [24]. A significant increase of the yield was also observed for the acetylated donor 4 by promoting the reaction with tin(IV) chloride. The use of this catalyst, unconventional in trichloroacetimidate chemistry, allowed to recover 3 in 76% with only traces of the 1-O-acetylated azidosphingosine byproduct 1a [23].
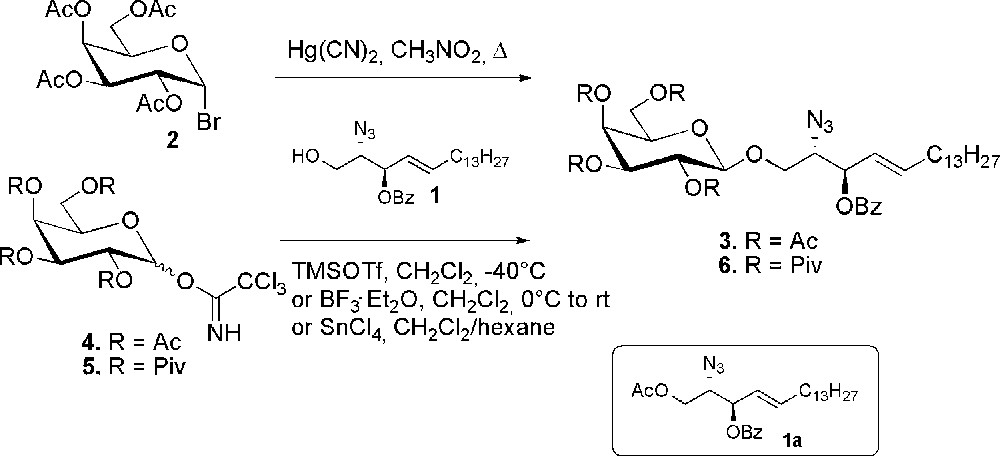
Glycosidation of 3-O-benzoyl azidosphingosine.
In carbohydrate chemistry, protecting groups play an important role both in terms of stereoselectivity of glycoside bond formation (α or β) and of the yield of such reactions. In this regard, the pivaloyl group has been found to be a very useful protecting group especially at C-2 to avoid orthoester formation and yet to give β-glycosylation. In the case of glycosylation of sphingoid bases a lower migration of the C-2 acyl protecting group to give 1a has also been observed, thus resulting in a more high yielding glycosylation. In fact, when the reaction was carried out on the pivaloyl derivative 5, the β-glycosylated 6 was obtained in excellent 88% yield [22e].
Similar procedures were then followed for the conversion of compounds 3 and 6 to final sulfatides. Thus, after deprotection of the sugar moiety and reduction of the azido group, the fatty acid chains were introduced and the resulting compounds were selectively sulfated at O-3 of the sugar moiety by using the stannylene methodology to afford the desired sulfatides. The non-physiological sulfatide 7, with a non-natural C-17 fatty acid chain, obtained starting from 3, has been prepared as an internal standard for the quantitative determination of urine sulfatides by mass spectrometry. Large quantities of sulfatides can be detected in the urine of patients with metachromatic leukodystrophy, so the search of sulfatiduria and the quantification of sulfatides appears an useful test for the follow-up of new therapies [24]. The family of sulfatides 8–11 differing in the acyl chain length has instead been obtained from 6. In particular, the sulfatides containing a palmitic, a stearic, a behenic or a nervonic fatty acid, all constituents of the natural mixture, have been prepared and subjected to immunological evaluation which showed that the activation of sulfatide-specific and CD1a-restricted T-cell clones is influenced by the acid chain length [22e].
Palmitic-containing sulfatide 8 could also be prepared from 3 with a different strategy [23], which does not exploit the stannylene methodology (Scheme 2). In this case, the acetates were removed from galactose and position 2 and 6 were benzoylated after introduction of the 3,4-O-isopropylidene acetal to give 12. The azido group was then reduced and N-acylated and the isopropylidene deprotected to compound 13. Introduction of the acetate in the axial position 4 was then followed by sulfation of the free 3-hydroxyl group. Final basic hydrolysis of the protecting groups gave sulfatide 8.This is a typical example of a standard carbohydrate protection-deprotection strategy, which underlines the great synthetic advantage introduced by the methodology of Flitch [20]. It is worth noting that the preparation of compound 8 was done in a wider context together with the synthesis of other sulfatide related compounds. The purpose was to evaluate their activity as ligands for selectine receptors. In fact, native sulfatide significantly blocks activity in selectine-dependent inflammatory response. The study showed a binding selectivity for the different position isomers of sulfatide.
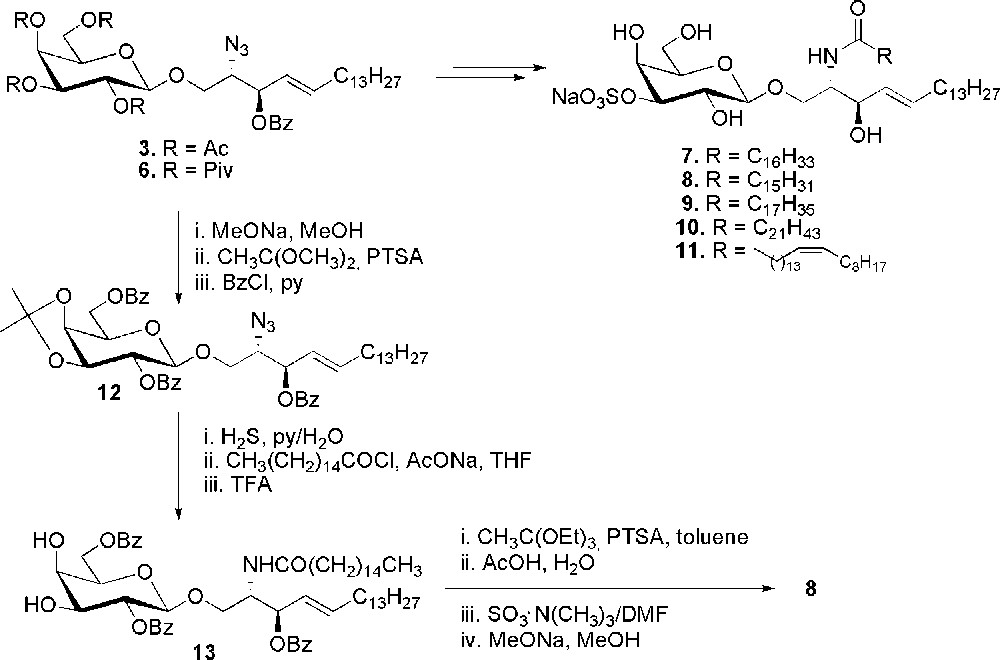
Synthesis of sulfatides 7–11.
Again, in the context of studies as selectine ligand, sulfatide was also obtained by glycosylation of ceramide [25] (Scheme 3). The galactosyl trichloroacetimidate donor 15 was prepared with a levulinoyl protecting group at position 3, orthogonal to the benzoates. Compound 15 was coupled with ceramide 16 in the presence of molecular sieves AW-300 and BF3·Et2O. β-glycoside was obtained in 74% yield and the levulinoyl group was selectively deprotected to give 17. Sulfatide 9 was recovered after sulfation and ester hydrolysis.
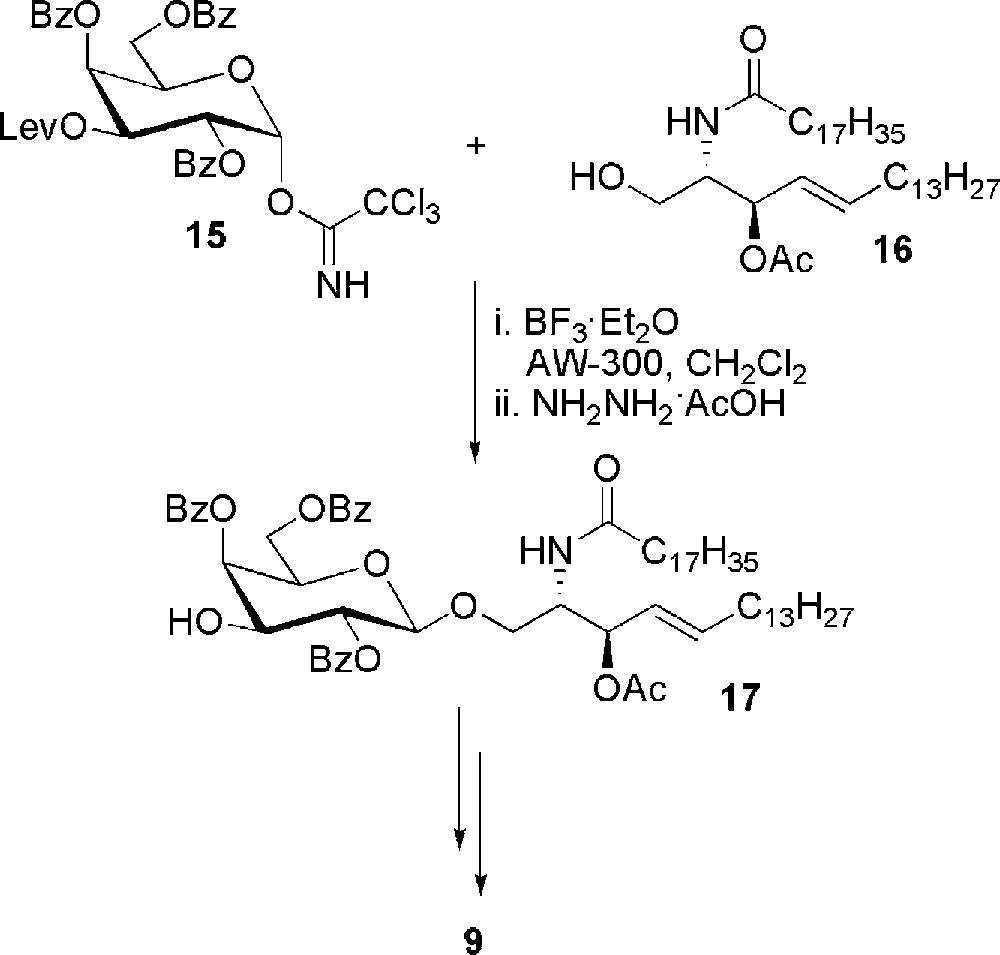
Synthesis of sulfatide 9 from ceramide 16.
More recently, a different glycosylation approach was used for the synthesis of the same sulfatide [26]. It is based on the glycosylation of sphingosine 18, where the amino group is doubly protected as tert-butoxycarbonyl and as N,O-isopropylidene with the secondary hydroxyl group (Scheme 4). The choice of this protecting group has been done in order to avoid the formation of the hydrogen bond between the electron pair of C-1 oxygen and the amide hydrogen of sphingosine, which in general reduces the reactivity of the sphingoid base towards glycosylation. Sphingosine 18 was glycosylated with peracetylated galactosylbromide 19, under the catalysis of Hg(II) salts. Galactosylsphingosine 20 was recovered in 62% yield. Higher yield (77%) and a cleaner reaction was obtained from the benzoylated donor 21. Starting from both 20 and 22 hydrolysis of the sphingosine protecting groups followed by N-acylation with stearic acid gave the deprotected galactocerebroside which was selectively sulfated to sulfatide 9.
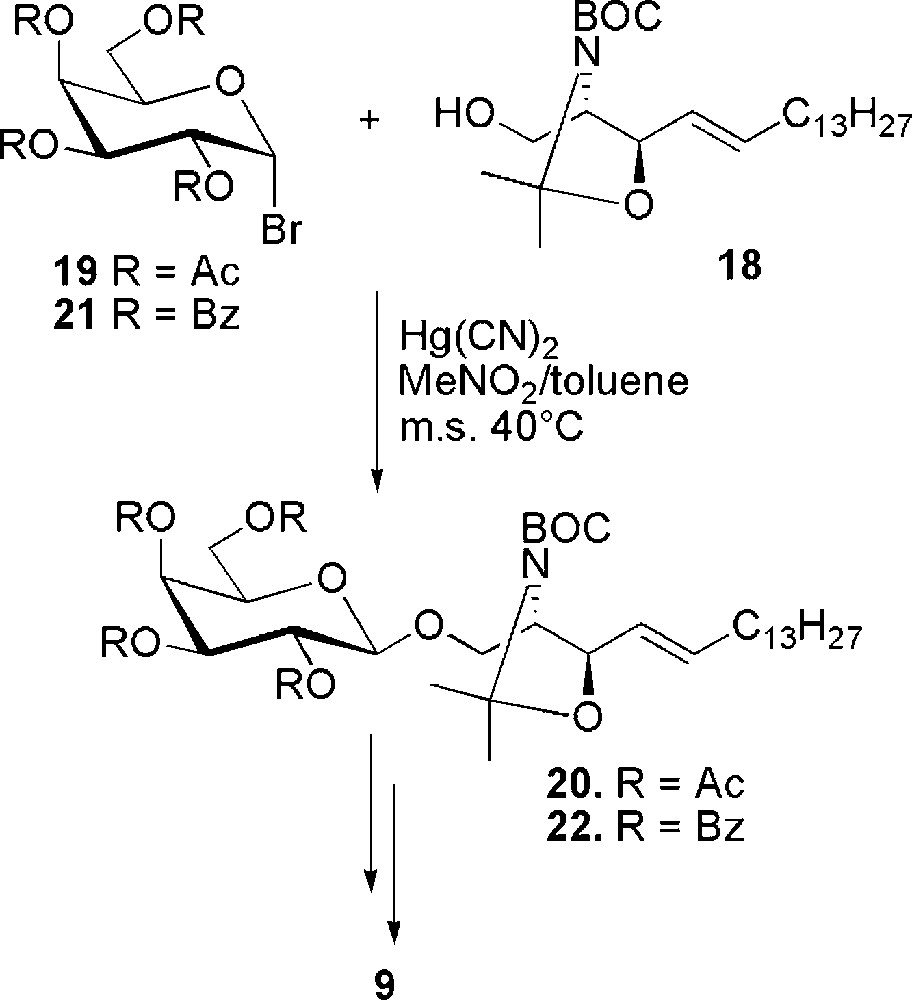
Synthesis of sulfatide 9 from doubly protected sphingosine 18.
3 Sulfatide related compounds modified in the saccharidic moiety
A significant contribution to the research has been given by the development of compounds structurally related to sulfatide. Herein, we will show only the derivatives that maintain the cerebroside feature, i.e. the β-d-galactosylceramide structure, with the characteristic presence of one sulfate, even if also analogues modified in the ceramide moiety have been reported [27]. Part of this work has been focused on the determination of the structural elements required for activity. This is particularly important when very precise chemical recognitions take place during the biological event and even minor structural modifications can influence the binding. For example, in the context of CD1 proteins, the activation of the immune system is a consequence of a specific trimolecular association between the glycolipid, the CD1 protein and the T-cell receptor. So, to assess the role of the position of the sulfate in CD1a-mediated sulfatide-specific T-cell activation, the synthesis of the 3 β-d-galactosyl ceramides 23–25 (Fig. 4) bearing a sulfate ester at C-2, C-4 or C-6 of galactose was carried out [28]. The compounds were synthesized by an orthogonal sulfation strategy from the common scaffold 26 (Scheme 5).
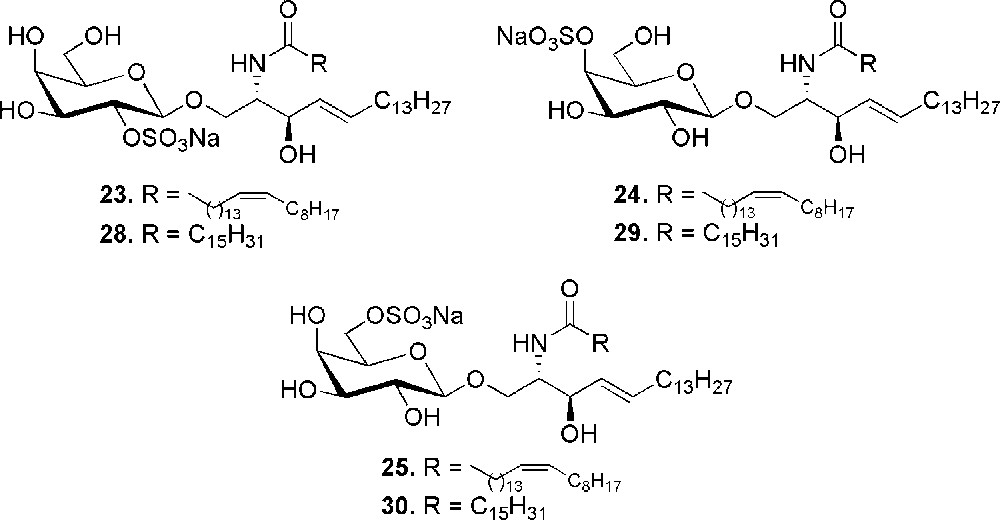
Position isomers of sulfatide.
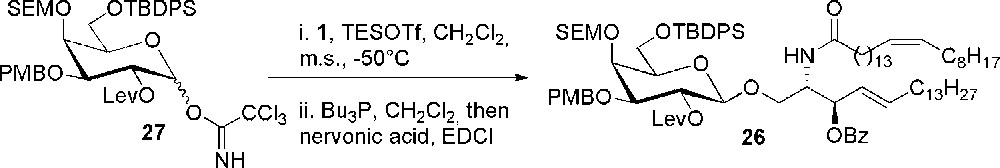
Synthesis of the fully orthogonally protected scaffold 26.
Compound 26 was obtained through an efficient glycosylation reaction catalyzed by TESOTf between the fully orthogonally protected galactosyl imidate 27 and 3-O-benzoyl azidosphingosine 1 in 86% yield, followed by conversion into ceramide. Each hydroxyl group of galactose was then selectively deprotected with respect to the other ones of galactose and to the one of ceramide and sulfated. Galactosyl ceramides 23, 24 and 25 were recovered after standard deprotection steps and subjected to immunological evaluation, which provided evidence of the influence of the sulfate position in the recognition event between sulfatide, the CD1 protein and the T-cell receptor.
Another family of position isomers of sulfatide, differing in the fatty acid residue (palmitic instead of nervonic), was also obtained in the frame of the above mentioned study of Marinier on the role of sulfatide as a ligand for P-selectin, which showed a binding selectivity for the position 2 and 3 of the sulfate group on the carbohydrate ring [23].
For this structure-activity relationship study, besides sulfatide 8 (Scheme 2), three other sulfated ceramides 28–30 were obtained by standard protection-deprotection strategies starting from 3. The coupling of the palmitic acid moiety at the beginning of the synthetic sequence was explored for the preparation of the 2-sulfated isomer 28 as depicted in Scheme 6. Thus, unprotected glycoside 31 was reduced, acylated and selectively protected at position 3, 4 and 6 of the sugar to give 32. Sulfation followed by protecting group cleavage gave galactocerebroside 28. Always starting from 31, selective sulfation of the 6-O position to 33 can be achieved after 3,4-O-isopropylidene formation. Compound 30 was in turn obtained after reduction, N-acylation and deprotection. A usual sequence of protection/deprotection steps on galactose, starting from 3, allowed to recover 34, which was the intermediate for the preparation of the 4-sulfated derivative 29. Compound 34 was sulfated after azido reduction and N-acylation and finally deprotected to sulfatide 29.
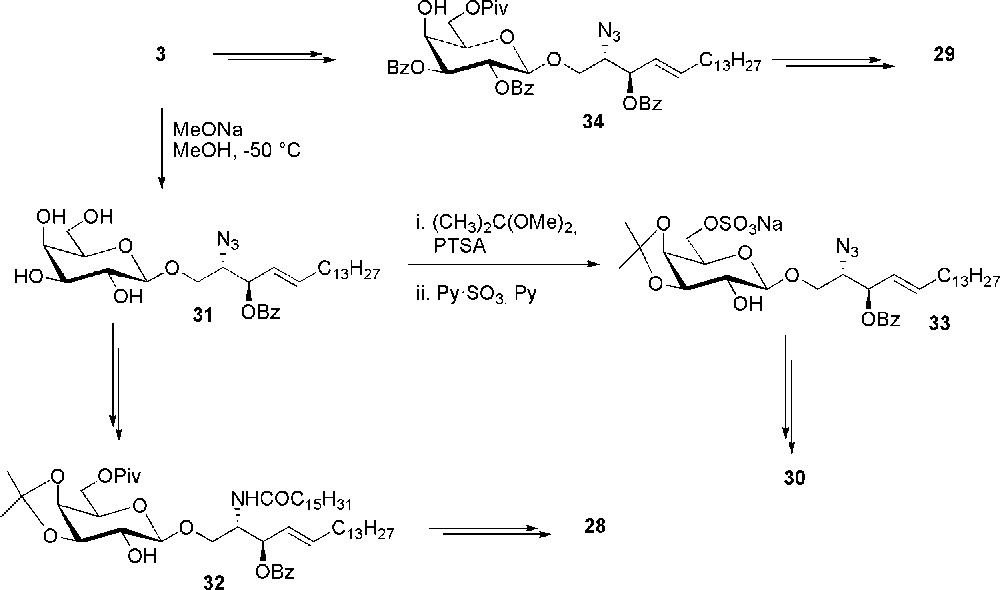
Synthesis of sulfatide isomers 28–30.
Another sulfatide related compound is the fluorescent glycolipid 35, prepared in order to evaluate its ability to be loaded on CD1a protein [29]. The hydrophobic binding region of CD1a is suited for the accommodation of the antigen alkyl chains. Thus, sulfatide 35 has been prepared with a dansyl group attached to the sugar and not to the ceramide part of the molecule and resulted a valuable tool for the study of sulfatide-CD1a binding properties. It was derived from glycolipid 36, in turn obtained in 55% yield by glycosylation, mediated by TESOTf, between donor 37 and 1 (Scheme 7). Most probably, the presence of a CH2COOMe group at position 6 of galactose, which has been introduced as an anchor point for the dansyl group, deactivates the glycosylation thus resulting in lower yields. Compound 36 was deprotected and the azidosphingosine moiety transformed into ceramide. The dansyl group was then attached through an ethylendiamine linker after hydrolysis of the methyl ester. Final selective sulfation gave the fluorescent sulfatide 35.
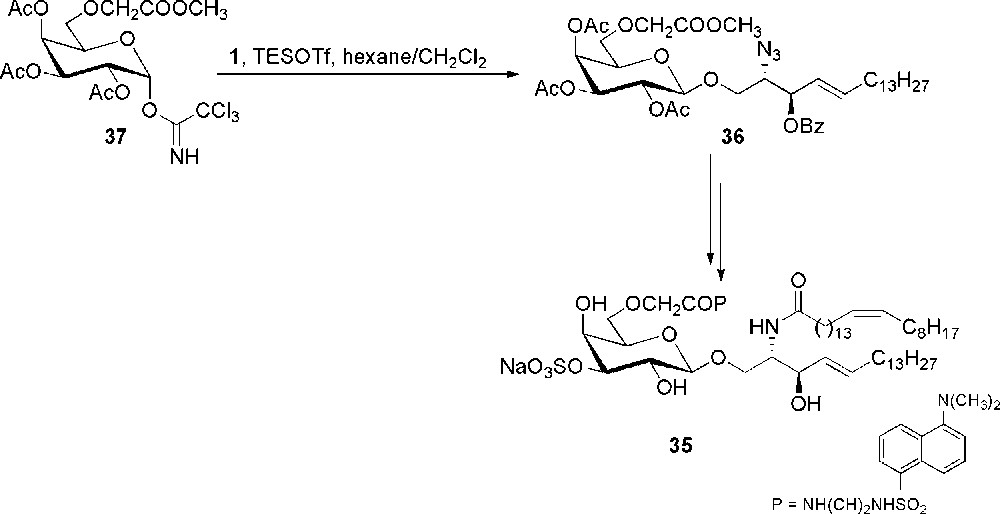
Synthesis of dansylated sulfatide 35.
Another modification that has been done on sulfatide has been to change the nature of the glycoside bond replacing the anomeric oxygen with a methylene group in order to limit the in vivo degradation of the glycolipid by glycosidases, and to test how this modification influences the activity with respect to that of the parent O-glycoside [30]. Thus, the C-glycoside analogue 38 of sulfatide was prepared starting from compound 39 and exploiting a [2,3]-Wittig sigmatropic rearrangement for the generation of the C-glycoside 40 (Scheme 8). Compound 40 was then converted into the β-ketophosphonate 41, which was the substrate for a Horner-Wadsworth-Emmons olefination for the introduction of the trans double bond and of the aliphatic chain. Stereoselective reduction of the keto-group followed by standards elaborations gave the final sulfatide derivative 38.
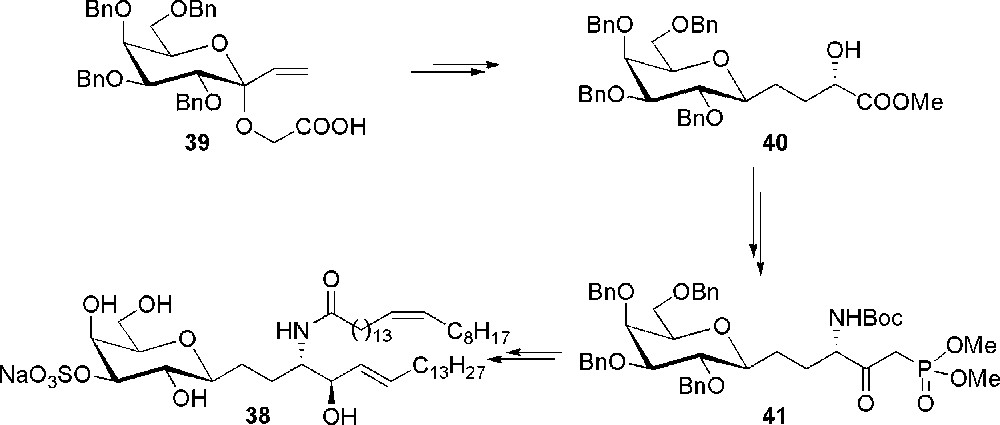
Synthesis of the C-sulfatide 38.
4 Conclusions
Sulfatide represents one of the major endogenous sulfated glycolipids. It exists in various isoforms with different physicochemical properties. Given its importance, this compound has largely stimulated the interest of the scientific community, as testified by the high number of literature reports. Nevertheless, in spite of the observation of a biological activity determined by a precise chemical structure, the syntheses of new derivatives and/or analogues have not been developed to a degree comparable to its biochemical importance. This account has the aim to collect what has been done in the synthesis of sulfatide and to become a stimulus for a renewed interest towards this glycolipid and the preparation of new analogues for biomedical use.
Acknowledgments
The authors gratefully thank the University of Milan and the University of Piemonte Orientale for financial support.