

1 Introduction
The vicinal haloazidation is an important organic transformation for simultaneous introduction of halogen and azide functionality onto an olefin. The resulting haloazides can be subjected for further synthetic manipulations to achieve a variety of compounds such as vinyl azide [1], amines [2], aziridins [3] and tetrazoles [4]. Vicinal iodoazidation has been known in the literature for a long time. Hantzsch reported for the first time in 1900 a strategy for the synthesis of 1,2-iodoazide using silver azide and iodine in ether [5]. 1,2-Iodoazides can also be synthesized by treating ICl/NaN3 [6], PhI(OAc)2/Et4NX/TMSN3 [7], CAN/NaI/NaN3 [8], IPy2BF4/TMSN3 [9] and Oxone/wet Al2O3/KI/NaN3 [10] with alkenes under various conditions. Poor selectivity and instability of these iodoazides are the main problems of these methods. Later, Hassner and Boerwinkle developed a procedure for vic- bromoazidation of alkene by addition of bromine azide generated from bromine and sodium azide [11]. This procedure requires the use of excess hydrazoic acid to suppress the formation of dibromo compounds. A similar procedure was also reported by Krief for bromoazidation of alkenes by bromine azide which was generated in situ from NBS and NaN3 [3]. Olah employed NBS and TMSN3 for the bromoazidation of alkenes in the presence of Nafion-H in DME/H2O [12]. However, this procedure was reported only for 1,2-disubstituted- and trisubstituted alkenes and there was no reaction with terminal alkenes. Recently, Hajra et al. reported a method for 1,2-bromoazidation of alkenes using NBS and TMSN3 in presence of Zn(OTf)2 catalyst [13,14], however, metal triflates are highly expensive. Although, this method works well with terminal and 1,2-disubstituted olefins, α, β−unsaturated carbonyl compounds take much longer time and require higher temperature to achieve good yield. Moreover, for chalcone, the reaction does not go into completion even after a long reaction time at high temperature. Bromoazidation with α, β−unsaturated carboxylic acid derivatives also takes much longer time even at higher temperature [14]. Therefore, the development of efficient methods for vicinal bromoazidation of alkenes, particularly in case of α, β–unsaturated alkenes, remains as a challenging task. We recently reported our preliminary result of our study on the vicinal bromoazidation of alkenes using TsNBr2 as bromine source and TMSN3 as azide source [15]. We are reporting herein a rapid and efficient procedure for vicinal bromoazidation of alkenes using NBS as bromine source and TMSN3 as azide source without any catalyst.
2 Results and discussion
In continuation of our work on iodine catalysed reaction [16], we intended to study the catalytic effect of iodine for bromoazidation reaction using NBS as a bromine source. The reason for choosing NBS is its commercial availability. It was reported that the metal triflate catalyzed bromoazidaiton reaction has limited scope in case of cinnamates and chalcones [13]. Therefore, we sought to develop an improved catalytic method for those particular substrates. We chose ethyl cinnamate as a model substrate to test the efficacy of iodine as catalyst for bromoazidaiton reaction (Scheme 1).
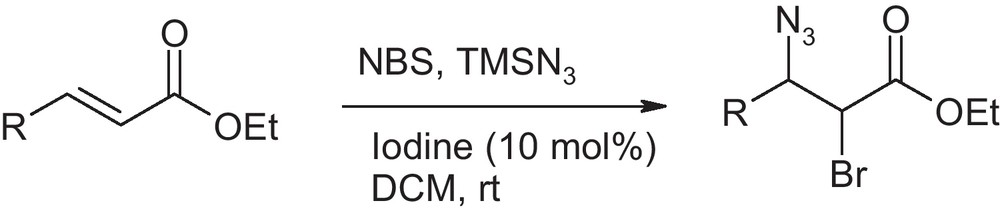
Initial experiments were performed in dichloromethane as solvent in presence of 10 mole % of iodine as catalyst. The reaction was carried out by adding iodine to a mixture of ethyl cinnamate, NBS and TMSN3. The reaction was complete in one hour with 85% yield (Table 1). In order to find out the best reaction condition, we investigated the effect of various solvents on the product yield. When acetonitrile was used as solvent, the reaction completes instantaneously to produce the corresponding bromoazide in 82% yield in the presence of iodine. Since the reaction rate was accelerated in acetonitrile, we reinvestigated the reaction in acetonitrile without the addition of catalytic iodine. Surprisingly, we found that the reaction works excellent with high yield without the catalyst. Thereafter, we tested again the reaction in DCM in absence of iodine. The reaction does not go into completion even after 18 hours in dichloromethane and the yield was poor (Table 1) in this case. We observed that the solubility of NBS in acetonitrile is very high as compared to DCM. This could be one of the reasons to drive the reaction forward.
Bromoazidation of ethyl cinnamate under different conditions.
| Entry | Solvent | Iodine (mol%) | Time | Yield (%) |
| 1 | CH2Cl2 | 10 | 1 h | 85 |
| 2 | Ether | 10 | 1 h | 42 |
| 3 | CH3CN | 10 | 1 min | 82 |
| 4 | CH3CN | 0 | 1 min | 84 |
| 5 | CH2Cl2 | 0 | 18 h | 40 |
After initial success in case of ethyl cinnamate, we extended the procedure to various cinnamates and chalcones (Scheme 2). The reaction was carried out by adding TMSN3 (1.1 equiv) to the solution of olefin in acetonitrile followed by the addition of NBS (1.2 equiv.). The whole process was carried out at room temperature under nitrogen atmosphere. We observed that the olefin disappears immediately after addition of NBS, which was confirmed by monitoring the reaction by TLC. The reaction mixture was stirred for another 10 minutes. After usual work-up the corresponding bromoazide was obtained in excellent yield. This process was extended to a variety of olefins in acetonitrile. The results are summarized in Table 2.
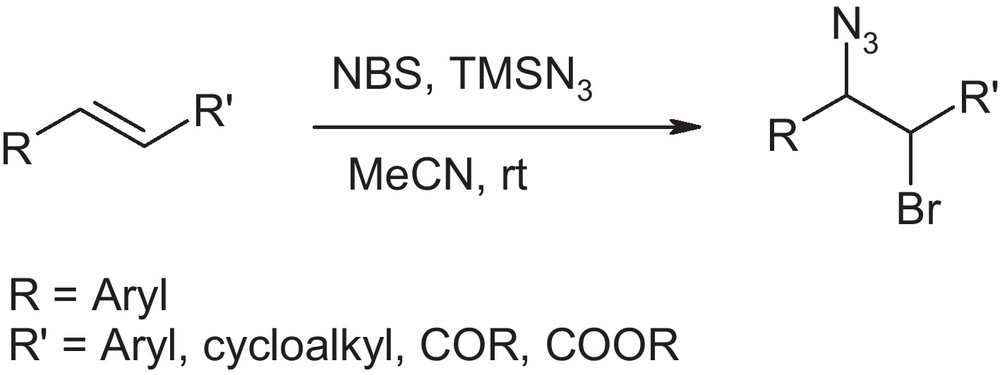
Bromoazidation of various alkenes with TsNBr2 and NBS.a
| Entry | Alkene (a) | Product (b) | Yieldb | anti/syn ratioc |
| 1 | 84 | 75:25 | ||
| 2 | 84 | 61:39 | ||
| 3 | 87 | 63:37 | ||
| 4 | 88 | -nd- | ||
| 5 | 89 | > 99.1 | ||
| 6 | 91 | > 99.1 | ||
| 7 | 92 | > 99:1 | ||
| 8 | 79 | − | ||
| 9 | 77 | − | ||
| 10 | 69 | 72:28 | ||
| 11 | 75 | > 99:1 |
a Reaction condition: olefin (1 mmol); TMSN3 (1.1 mmol); NBS(1.2 mmol); MeCN (2 mL); rt, 10 minutes.
b isolated yield after chromatographic purification; characterized by IR, NMR and Mass spectral data.
c anti/syn ration was determined from NMR spectral data; nd: not determined.
It is seen from Table 2 that various cinnamic esters reacted successfully to give the corresponding 1,2-bromoazide in high yields. The reaction is regioselective, however, moderate steroselectivity was obtained for most of the cases. Bromoazides produced from cinnamic esters have anti/syn ratio of about 3:1 which was determined from the 1H NMR analysis. The reaction also works excellent in case of chalcone as well as stillbene with exclusive antiselectivity (Table 2, entry 5–7). After being successful in case of cinammic esters, stilbene and chalcone, we extended the procedure to styrenes, 1,2-dihydronaphthalene and cyclohexene, which produced the corresponding bromoazide in high yield. We did not observe the formation of vic-dibromo adduct of styrene [13].
3 Conclusions
In conclusion, an efficient protocol for vicinal bromoazidation of olefins has been established by using N-bromosuccinimide and trimethylsilyl azide as the bromine and azide sources respectively in acetonitrile at room temperature without the use of any catalyst. The procedure is very fast, easy to perform and applicable to various olefins such as cinnamates, chalcone, and styrenes and cyclohexene to give corresponding 1, 2-bromoazide in high yield.
4 Experimental
4.1 General
All the chemicals used were of analytical grade. IR spectra were recorded by using Perkin Elmer Spectrum RX I FT-IR Spectrometer. Mass spectra were recorded on a Perkin Elmer Clarus 600 C mass spectrometer. 1H and 13C NMR spectra were obtained in CDCl3 at 300 and 75 MHz respectively, using Bruker Ultrashild 300 MHz spectrometer (TMS as internal reference). Some of the 1H and 13C spectra were obtained in CDCl3 at 400 and 100 MHz respectively, using Bruker Avance 400 MHz NMR Spectrometer.
4.2 Synthetic procedures
To a solution of olefin (1 mmol) in acetonitrile (2 ml) TMSN3 (1.1 mmol), and then NBS (1.2 mmol) were added under nitrogen atmosphere and stirred. After completion of the reaction sodium thiosulfate (200 mg approx.) was added and the reaction mixture was stirred for 15 minutes. The reaction mixture was taken up in ethyl acetate, dried over anhydrous sodium sulfate, and concentrated. The crude product was purified by flash chromatography on silica gel (230–400 mesh) with petroleum ether/ethyl acetate (98:2) as eluent.
Ethyl 3-azido-2-bromo-3-phenyl propanoate (1b). IR (KBr, cm−1) ν: 2106 (N3), 1743, 1265, 1022, 740; 1H NMR (CDCl3, 400 MHz) δ: 7.34- 7.26 (m, 5H), 5.14 (d, 1H, J = 9.6 Hz), 4.33 (d, 1H, J = 9.6 Hz), 4.24-4.20 (m, 2H), 1.26 (t, 3H, J = 7.6 Hz); 13C NMR (CDCl3, 100 MHz) δ: 167.5, 136.7, 129.4, 128.9, 128.2, 67.2, 62.4, 49.4, 14.0; GCMS (M/Z %): 269 (M+- 28, N2 elimination, 61.5), 271 (61.5), 195 (61.5), 197 (59), 189 (33.33), 162 (25.64), 143 (58.97), 116 (87.18), 90 (100), 63 (43.59), 29 (76.9).
Methyl 3-azido-2-bromo-3-phenyl propanoate (2b). IR (KBr, cm−1) ν: 3048, 2950, 2116 (N3), 1748, 1611, 1440, 1381, 1265, 742; 1H NMR (CDCl3, 400 MHz) δ: 7.48- 7.35 (5H, m), 5.22 (d, 1H, J = 8 Hz), 4.44 (d, 1H, J = 8 Hz), 3.85 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ: 168.0, 136.7, 130.4, 128.8, 127.6, 67.1, 52.9, 49.0; GCMS (M/Z%): 255(M+- 28, N2 elimination, 40.8), 257 (39.5), 195 (43.75), 175 (24.58), 143 (48.75), 117 (100), 89 (95.4), 63(32.91), 28 (48.75).
Methyl 3-azido-2-bromo-3-(4-chlorophenyl) propanoate (3b). IR (KBr, cm−1) ν: 3048, 2950, 2106, 1749, 1594, 1491, 1438, 1264, 1204, 1094, 1015, 832, 738, 523; 1H NMR (CDCl3, 400 MHz) δ: 7.42- 7.31 (m, 4H), 5.19 (d, 1H, J = 8 Hz), 4.405 (d, 1H, J = 12 Hz), 3.83 (s, 3 H); 13C NMR (CDCl3, 100 MHz) δ: 167. 7, 135.1, 131.4, 129.6, 129.1, 67.1, 52.9, 48.3; GCMS (M/Z%): 289 (M+-28, N2 elimination, 45.83), 291 (56.67), 231(59.16), 151(100), 123 (45.41), 89 (84.58), 63 (28.33), 28 (43.75).
Methyl 3-azido-2-bromo-3-(4-methoxyphenyl) propanoate(4b). IR (KBr, cm−1) ν: 3067, 3028, 2950, 2852, 2106 (N3), 1748, 1719, 1617, 1493, 1438, 1376, 1327, 1264, 1206, 1086, 1021, 769, 698; 1H NMR (CDCl3, 400 MHz) δ: 7.53-6.90 (m, 4H), 4.87 (d, 1H, J = 12 Hz), 4.23 (d, 1H, J = 12 Hz), 3.88 (s, 3 H), 3.84 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ: 167.3, 156.4, 132.4, 128.1, 111.6, 66.4, 56.1, 52.8, 45.8; GCMS (M/Z %): 268 (M+-28, N2 elimination, 4), 270 (4), 226 (16.67), 119 (15), 147 (32.5), 132 (16.25), 104 (14.58), 28 (100).
3- Azido-2-bromo-1,3-diphenylpropan-1-one (5b). IR (KBr, cm−1) ν: 3048, 2930, 2852, 2440, 2096 (N3), 1689, 1648, 1592, 1494, 1440, 1366, 1327, 1302, 1253, 1199, 1214, 1175, 1027, 983, 885, 777; 1H NMR (CDCl3, 300 MHz) δ: 8.15-8.09 (m, 2 H), 7.77-7.43 (m, 8 H), 5.47 (d, 1H, J = 10.2 Hz), 5.19 (d, 1H, J = 10.5 Hz); 13C NMR (CDCl3, 75 MHz) δ: 193.3, 137.1, 135.2, 134.4, 130.1, 129.4, 129.0, 128.9, 128.2, 64.3, 48.7.
3-Azido-2-bromo-3-(4-chlorophenyl)-1-phenylpropan-1-one (6b). IR (KBr, cm−1) ν: 3058, 2979, 2116 (N3), 1652, 1422, 1264, 1094, 895, 748; 1H NMR (CDCl3, 300 MHz) δ: 8.07 (d, 2H, J = 7.5 Hz), 7.78-7.27 (m, 7 H), 5.43 (d, 1H, J = 10.2 Hz); 13C NMR (CDCl3, 75 MHz) δ: 193.0, 135.8, 135.3, 135.1, 134.5, 129.6, 129.3, 129.1, 128.9, 64.3, 47.6.
1-Azido-2-bromo-1,2-diphenylethane (7b). IR (KBr, cm−1) ν: 2920, 2106 (N3), 1264, 739, 700; 1H NMR (CDCl3, 300 MHz) δ: 7.42-7.11 (m, 10H), 5.06 (d, 1H, J = 5.7 Hz), 4.99 (d, 1H, J = 9.3 Hz); 13C NMR (CDCl3, 75 MHz) δ: 138.1, 136.2, 128.6, 128.5, 128.4, 128.2, 127.7, 127.5, 57.9, 55.6.
1-(1-azido-2-bromoethyl) benzene (8b). IR (KBr, cm−1) ν: 2096 (N3), 1425, 1265, 741; 1H NMR (CDCl3, 400 MHz) δ: 7.47-7.38 (m, 5H), 5.28- 4.79 (m, 1H), 4.13-3.41 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ: 138.4, 128.9, 128.8, 127.5, 57.3, 51.0; GCMS (M/Z %): 197 (M+ - 28, N2 elimination, 47), 199 (41.66), 118 (97), 91(100), 58(47), 39 (17.6), 28 (17.6),18 (20.6).
1-(1-azido-2-bromoethyl)-4-chlorobenzene (9b). IR (KBr, cm−1) ν: 2920, 2508, 2106, 1596, 1491, 1260, 1094, 1013, 828, 728, 654; 1H NMR (CDCl3, 400 MHz) δ: 7.41-7.37 (m, 4H), 4.99-4.95 (m,1H), 3.93-3.79 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ: 136.9, 134.9, 129.2, 128.2, 65.3, 49.8.
1- Azido-2-bromocyclohexane (10b). IR (KBr, cm−1) ν: 2930, 2852, 2096 (N3), 1994, 1739, 1698, 1650, 1538, 1516, 1456, 1395, 1262, 1189, 1086, 1023; 1HNMR (CDCl3, 400 MHz) δ:3.90-3.81 (m, 1H), 3.50-3.44 (m, 1H), 1.95-1.71 (m, 4H), 1.43-1.30 (m, 4H); 13C NMR (CDCl3, 100 MHz) δ: 66.4, 54.9, 36.3, 31.8, 25.6, 23.6.
1-Azido-2-bromo-1, 2, 3, 4-tetrahyonaphthlene (11b). IR (KBr, cm−1) ν: 2923, 2357, 2100, 1603, 1491, 1451, 1249, 1204, 812, 748; 1H NMR (CDCl3, 400 MHz) δ:7.25-7.11 (m, 4 H), 4.67 (d, 1H, J = 4.8 Hz), 4.46 - 4.38 (m, 1H), 3.04- 2.95 (m, 1H), 2.84-2.77 (m, 1H), 2.44 -2.31 (m, 1H), 2.21 -2.11 (m, 1H); 13C NMR (CDCl3, 100 MHz) δ:135.4, 130.5, 129.7, 129.2, 128.739, 126. 7, 65.1, 50.4, 27.8, 25.9; GCMS (M/Z %): 223 (M + -28, N2 elimination) 225 (10.8), 144 (100), 129 (20.1), 117 (40), 90 (20.5), 63 (10.6), 51 (10.2), 39 (10.9), 28 (60.8).
Acknowledgements
Financial Support from DST (Grant No. SR/S1/RFPC-07/2006) is gratefully acknowledged. IS thanks to CSIR for research fellowship.
Vous devez vous connecter pour continuer.
S'authentifier