

1 Introduction
1,2,3-triazoles are important nitrogen heterocyles which display many interesting properties including antibacterial [1], herbicidal, fungicidal [2], antiallergic [3], anti-HIV [4], GSK-3 inhibiting [5], and antineoplastic activities [6]. Moreover, they have been widely used in various research fields, such as biological [7], and material sciences [8], medicinal [9], and synthetic organic chemistry [10].
One-pot multi-step reactions have a number of advantages including minimizing waste, saving energy, and reducing operating times. This process is especially useful when the reaction intermediates are unstable and difficult to isolate. Recently, efforts have been developed for the synthesis of 1,2,3-triazoles by copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC) using a multi-step, one-pot synthetic approach [11]. Copper(I)-catalyzed 1,3-cycloaddition reaction often referred to as “click-chemistry” [12] offered an efficient route to the synthesis of 1,4-disubstituted-1,2,3-triazoles. Also the Ru-catalyzed reaction of terminal alkynes with alkyl azides was discovered to be a unique way to synthesize 1,5-disubstituted-1,2,3-triazoles [13]. In these methods inorganic azides, NaN3 and KN3 are not active enough under mild conditions and it is inconvenient to prepare 4,5-disubstituted 1,2,3-(NH)-triazoles. Only a few methods for the synthesis of 4,5-disubstituted 1,2,3-(NH)-triazoles have been reported [14]. It is therefore necessary to develop novel ways of synthesizing 4,5-disubstituted 1,2,3-(NH)-triazoles.
2 Results and discussion
Some new approachs for constructing 4,5-disubstituted 1,2,3-(NH)-triazoles are based on Sonogashira coupling reaction [15]. These methods are efficient, but they have several drawbacks, which include the requirements of expensive palladium catalyst, dry solvents, and inert atmosphere.
In this article, we report an efficient, one-pot, copper and palladium free method for the synthesis of 4,5-disubstituted 1,2,3-(NH)-triazoles 4 via cross coupling reaction/1,3-dipolar cycloaddition of acid chlorides 1, terminal alkynes 2 and sodium azide in the presence of silica supported-zinc bromide under aerobic conditions (Scheme 1).
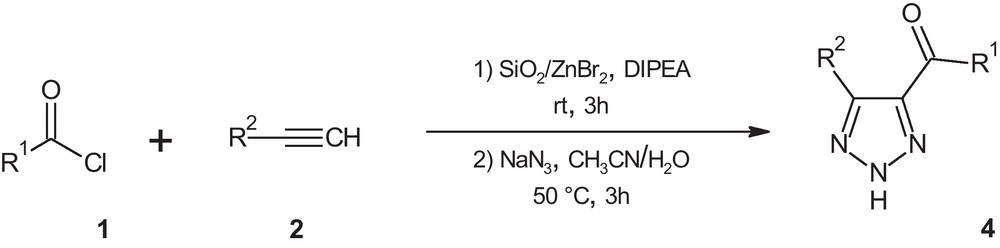
Synthesis of 4,5-disubstituted 1,2,3-(NH)-triazoles from various acid chlorides, terminal alkynes and sodium azide in the presence of silica supported-zinc bromide under aerobic conditions.
Recently we reported the synthesis of ynones by the cross-coupling reaction of acid chlorides with terminal alkynes in the presence of silica-supported zinc bromide (ZnBr2/SiO2) under solvent free conditions [16]. We decided to use the same procedure followed by 1,3-dipolar cycloaddition for the synthesis of 4,5-disubstituted 1,2,3-(NH)-triazoles.
In our starting experiment, phenyl acetylene, 4-chlorobenzoyl chloride, sodium azide were put in one-pot in the presence of ZnBr2 (1.2 eq) and a base (DIPEA or Et3N) in various solvents (CH3CN, dioxane, THF, DMF) was added to the mixture and the reaction was carried out at room temperature for 10 h, after which the temperature was raised to 50 °C and the reaction was continued for a further 5 h. Unfortunately, only a very poor yield of the desired product were formed. But when the reaction of phenyl acetylene and 4-chlorobenzoyl chloride were carried out using ZnBr2 (1.2 eq) or ZnBr2/SiO2 (10 mol%) catalyst in various solvents or under solvent free condition at room temperature, in advance, followed by sodium azide 1,3-dipolar cycloaddition at 50 °C, (4-bromophenyl)(5-phenyl-2H-1,2,3-triazol-4-yl)methanone, 4c was obtained in 62–96% isolated yield (Table 1). Optimization of the conditions was reached when ZnBr2/SiO2 (10 mol%) was used as the catalyst under solvent free condition at room tempreture followed by sodium azide cycloaddition in CH3CN/H2O (3:1) at 50 °C (Table 1, entry 7).
Silica supported-zinc bromide coupling reaction of p-chlorobenzoyl chloride 1b with phenylacetylene followed by sodium azid 1,3-dipolarcycloaddition in the presence of different bases and solventsa.
| Entry | Solvent | Base | Catalyst | Cyloaddition solvent | Yield (%) |
| 1 | CH3CN | DIPEAb | ZnBr2c | CH3CN/H2O | 76 |
| 2 | CH3CN | Et3N | ZnBr2 | CH3CN/H2O | 62 |
| 3 | DMF | Et3N | ZnBr2 | DMF/H2O | 78 |
| 4 | DMF | DIPEA | ZnBr2 | DMF/H2O | 85 |
| 5 | CH3CN | DIPEA | SiO2/ZnBr2e | CH3CN/H2O | 80 |
| 6 | DMF | DIPEA | SiO2/ZnBr2 | DMF/H2O | 71 |
| 7 | –d | DIPEA | SiO2/ZnBr2 | CH3CN/H2O | 96 |
| 8 | –d | Et3N | SiO2/ZnBr2 | CH3CN/H2O | 74 |
| 9 | –d | DIPEA | SiO2/ZnBr2 | H2O | 90 |
| 10 | –d | DIPEA | SiO2/ZnBr2 | DMSO/H2O | 70 |
a Reaction conditions: p-chlorobenzoyl chloride (1.2 mmol), phenylacetylene (1.0 mmol), base (1.2 mmol), solvent (1 mL) and catalyst, stir room temperature (3 h), followed by sodium azid (1.2 mmol) addition in solvent, 50 °C (3 h).
b Diisopropylethylamine.
c 1.2 eq.
d Without solvent.
e 10 mol%.
In order to explore the scope and generality of this protocol, we examined various benzoyl chlorides and different 1-alkynes using the optimized procedure (Table 2). The results show that this reaction is equally facile with both electron-donating and electron-withdrawing substituents present on the benzoyl chloride, resulting in good-to-high yields of the corresponding triazoles. Aliphatic terminal alkynes also reacted smoothly, affording the desired product in 88–95% yield. The structure of compounds was established by spectroscopic data and elemental analysis.
Synthesis of 4,5-disubstituted 1,2,3-(NH)-triazoles in the presence of silica supported-zinc bromidea.
| Entry | R1 | R2 | Product | Mp (°C) | Yieldb (%) | Ref |
| 1 | C6H5 | C6H5 | 4a | 114–116 | 92 | [15a] |
| 2 | C6H5 | C3H7 | 4b | 85–87 | 94 | [15a] |
| 3 | 4-BrC6H4 | C6H5 | 4c | 92–94 | 95 | New |
| 4 | 4-ClC6H4 | C6H5 | 4d | 131–133 | 96 | [15a] |
| 5 | 2-ClC6H4 | C3H7 | 4e | Oil | 92 | New |
| 6 | 2-ClC6H4 | C6H5 | 4f | Oil | 91 | [15a] |
| 7 | 4-ClC6H4 | C3H7 | 4g | 75–77 | 93 | New |
| 8 | 4-MeC6H4 | C6H5 | 4h | 117–119 | 90 | [15a] |
| 9 | 2-MeC6H4 | C6H5 | 4i | 89–91 | 87 | [15a] |
| 10 | 2-MeC6H4 | C3H7 | 4j | Oil | 88 | New |
| 11 | 4-ClC6H4 | C6H13 | 4k | Oil | 94 | New |
| 12 | 4-NO2C6H4 | C6H5 | 4l | 150–152 | 82 | [15a] |
| 13 | 2,4-Cl2C6H4 | C6H5 | 4m | 68–70 | 96 | New |
| 14 | 4-ClC6H4 | C4H9 | 4n | 73–75 | 95 | [15a] |
a Reaction conditions: benzoyl chloride (1.2 mmol), terminal alkyne (1.0 mmol), base (1.2 mmol), SiO2/ZnBr2 (10 mol%), stir room temperature (3 h), followed by addition of sodium azid (1.2 mmol), 50 °C (3 h).
b Isolated yield.
The proposed mechanism for the steps involved in the reactions is shown in Scheme 2. The key step of the reaction is the cross-coupling reaction of terminal alkynes with benzoylchlorides to afford the ynone intermediate 3. Subsequent 1,3-dipolar cycloaddition with sodium azide completes the reaction mechanism.
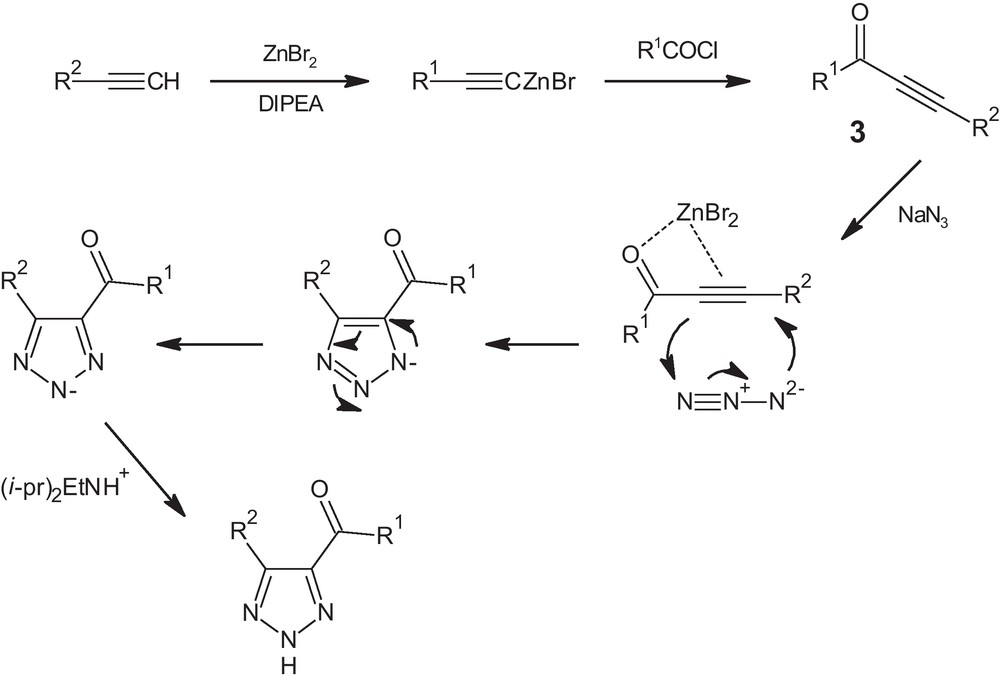
Proposed mechanism for the formation of 4,5-disubstituted-1,2,3-(NH)-triazoles, from acyl chlorides, terminal alkynes and sodium azide in the presence of silica supported-zinc bromide.
3 Conclusion
In conclusion, we have developed an efficient, facile, convenient, one-pot, copper and palladium free method for the synthesis of 4,5-disubstituted 1,2,3-(NH)-triazoles via cross-coupling reaction/1,3-dipolar cycloaddition of acid chlorides, terminal alkynes and sodium azide in the presence of silica supported-zinc bromide in aerobic conditions.
4 Experimental
All the reagents used were of general reagent grade. IR spectra were obtained as potassium bromide pellets or solvent in the range of 400–4000 cm−1 on a Shimadzu Model 460 spectrometer. 1H NMR spectra were recorded on a Brucker BRX 400 AVANCE spectrometer. Elemental analyses were performed on a Thermo Finnigan Flash EA microanalyzer.
4.1 Preparation of silica-supported zinc bromide
Silica gel (Wakogel C-100, 3.65 g) was added to a solution of ZnBr2 (6 mmol, 1.35 g) in EtOH (20 ml), and the mixture was heated at reflux for 1 h. The solvent was removed on a rotary evaporator, and the product was dried under vacuum at 150 °C for 10 h. Inductively coupled plasma atomic absorption spectrometry (ICP) indicated that 1.2 mmol of ZnBr2 was supported on 1 g of ZnBr2/SiO2.
4.2 General procedure for the preparation of 4,5-disubstituted 1,2,3-(NH)-triazoles (3a-n)
A test-tube was charged with the acyl chloride (1.2 mmol), a terminal alkyne (1.0 mmol), ZnBr2/SiO2 (0.1 g, 0.12 mmol), and DIPEA (1.2 mmol), and the mixture was stirred at room temperature for 3 h under anhydrous conditions. Upon completion of the coupling reaction (monitored by TLC), sodium azide (1.2 mmol) in CH3CN/H2O (3:1) (2 ml) was added and and the mixture was heated at 50 °C with stirring for a further 3 h. After completion of the reaction (monitored by TLC), the solvent was evaporated and residue extracted with CH3CN. Concentrating the solution gave the crude product which was subjected to column chromatography using CHCl3–CH3OH (98:2) as eluent to obtain an analytically pure product. Characterization data for new compounds are given below.
4.3 (4-bromophenyl)(5-phenyl-2H-1,2,3-triazol-4-yl)methanone (4c)
1H NMR (400 MHz, CDCl3): δ7.85 (d, J = 8.0 Hz, 2H, ArH), 7.72 (d, J = 8.0 Hz, 2H, ArH), 7.67–7.47 (m, 5H, ArH), 5.77 (s, 1H, NH); 13C NMR (100 MHz, CDCl3): δ 188.52, 132.72, 132.41, 132.20, 131.56, 130.53, 130.00, 129.93, 129.62, 129.41, 128.60. IR (KBr): 3290 (NH), 1640 (CO), 1590, 1560, 1490, 740 cm−1; Anal. Calcd for C15H10BrN3O: C, 54.90; H, 3.07; N, 12.80; Found: C, 54.88; H, 3.13; N, 12.78.
4.4 (2-chlorophenyl)(5-propyl-2H-1,2,3-triazol-4-yl)methanone (4e)
1H NMR (400 MHz, DMSO-d6): δ7.90 (d, J = 7.4 Hz, 1H, ArH), 7.46–7.75 (m, 3H, ArH), 6.82 (s, 1H, NH), 2.71 (t, J = 7.4 Hz, 2H, CH2), 1.52 (m, 2H, CH2), 0.98 (t, J = 7.6 Hz, 3H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 187.61, 152.69, 142.74, 138.34, 136.85, 133.57, 131.20, 129.14, 127.82, 30.23, 20.66, 13.42. IR (KBr): 3300 (NH), 2920, 1680 (CO), 1590, 1420, 1320, 760 cm−1; Anal. Calcd for C12H12ClN3O: C, 57.72; H, 4.84; N, 16.83; Found: C, 57.70; H, 4.85; N, 16.85.
4.5 (4-chlorophenyl)(5-propyl-2H-1,2,3-triazol-4-yl)methanone (4g)
1H NMR (400 MHz, DMSO-d6): δ 8.10 (d, J = 8.3 Hz, 2H, ArH), 7.50 (d, J = 8.4 Hz, 2H, ArH,), 6.50 (s, 1H, NH), 2.94 (t, J = 7.6 Hz, 2H, CH2), 1.60 (m, 2H, CH2), 1.00 (t, J = 7.5 Hz, 3H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 187.26, 153.70, 141.81, 139.56, 134.38, 131.82, 129.04, 30.22, 20.64, 13.55. IR (KBr): 3285 (NH), 2950, 1678 (CO), 1588, 1425, 1310, 750 cm−1; Anal. Calcd for C12H12ClN3O: C, 57.72; H, 4.84; N, 16.83; Found: C, 57.73; H, 4.83; N, 16.84.
4.6 (2-methylphenyl)(5-propyl-2H-1,2,3-triazol-4-yl)methanone (4j)
1H NMR (400 MHz, DMSO-d6): δ 7.66 (d, J = 7.7 Hz, 1H, ArH), 7.20–7.45 (m, 3H, ArH), 2.61 (t, J = 7.7 Hz, 2H, CH2), 2.22 (s, 3H, CH3), 1.68 (m, 2H, CH2), 0.83 (t, J = 7.4 Hz, 3H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 185.02, 152.80, 141.77, 140.23, 137.33, 132.58, 132.43, 131.62, 125.76, 29.88, 20.64, 20.33, 13.67. IR (KBr): 3300 (NH), 2950, 1640 (CO), 1540, 1445, 1380, 1240, 840, 750 cm−1; Anal. Calcd for C13H15N3O: C, 68.10; H, 6.59; N, 18.33; Found: C, 68.18; H, 6.61; N, 18.35.
4.7 (4-chlorophenyl)(5-hexyl-2H-1,2,3-triazol-4-yl)methanone (4k)
1H NMR (400 MHz, DMSO-d6): δ 7.82 (d, J = 8.3 Hz, 2H, ArH), 7.35 (d, J = 8.3 Hz, 2H, ArH), 2.62 (t, J = 6.8 Hz, 2H, CH2), 1.58 (m, 2H, CH2), 1.20 (m, 4H, 2CH2), 0.82 (t, J = 6.4 Hz, 3H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 187.26, 153.65, 142.00, 139.79, 134.48, 131.71, 128.93, 31.75, 30.85, 29.40, 27.52, 23.18, 14.13; IR (KBr): 3300 (NH), 2900, 1690 (CO), 1580, 1450, 1380, 1258, 750 cm−1; Anal. Calcd for C15H18ClN3O: C, 61.75; H, 6.22; N, 14.40; Found: C, 61.73; H, 6.23; N, 14.38.
4.8 (2,4-dichlorophenyl)(5-phenyl-2H-1,2,3-triazol-4-yl)methanone (4m)
1H NMR (400 MHz, DMSO-d6): δ 9.23 (s, 1H, NH), 8.14 (d, J = 8.5 Hz, 2H, ArH), 7.68 (s, 1H, ArH), 7.57–7.26 (m, 5H, ArH); 13C NMR (100 MHz, DMSO-d6): δ 185.92, 153.37, 138.65, 138.08, 134.48, 133.82, 131.00, 130.60, 130.45, 129.35, 128.45, 128.13, 127.54; IR (KBr): 3293 (NH), 2900, 1648 (CO), 1590, 1540, 1380, 1320, 820, 780 cm−1; Anal. Calcd for C15H9Cl2N3O: C, 56.63; H, 2.85; N, 13.21; Found: C, 56.65; H, 2.83; N, 13.22.