

1 Introduction
The search for a more efficient and environmentally benign synthetic pathway is always a challenge for chemists. The synthesis of 1,3-diphenyl-2-propenone (chalcone) is drawing attention because of its versatility and physiological activity [1–10] such as antimicrobial, antioxidant, antimalarial, anti-inflammatory, antileishmanial and anticancer. Apart from its biological activity these substrates are widely used in various organic reactions like flavonoid, epoxide synthesis [11,12]. The typical laboratory procedure uses bases like NaOH, KOH, Ba(OH)2, etc. as catalyst in the synthesis of chalcones from benzaldehyde and acetophenone or their derivatives [13–15]. The acid-catalyzed synthesis of chalcones includes AlCl3, HCl, BF3-Et2O, TiCl4 and RuCl3, etc. Palleros has recently reported [16] the synthesis of chalcones under solvent-free condition where heating under reflux was replaced by the grinding of benzaldehyde and acetophenone with mortar and pestle. Functionalized aminopropylated nanosilica catalysts were also investigated in the Claisen–Schmidt preparation of chalcones under solvent-free conditions [17]. The condensation between acetophenone and benzaldehyde to produce chalcones using acidic ionic liquids with dual purpose as catalyst and solvent was consciously designed by Liu et al. [18]. A conventional sonochemical method [19] was also attempted in the synthesis of chalcone at room temperature to establish the reaction as an energy saving synthesis. However, many of these synthetic routes are associated with several consequences like more reaction time, low yield, difficult work-up procedure, waste disposal problems etc.
In this report, we are presenting a method for the synthesis of chalcones in a solvent-free condition under microwave irradiation in presence of chlorotrimethylsilane (CTMS) as catalyst (Scheme 1). The primary objectives of this investigation are:
- • minimize reaction time;
- • solvent-free green approach;
- • easy work-up procedure;
- • use of microwave reactor;
- • increase productivity and;
- • antimicrobial activity studies in search of promising drug candidate.
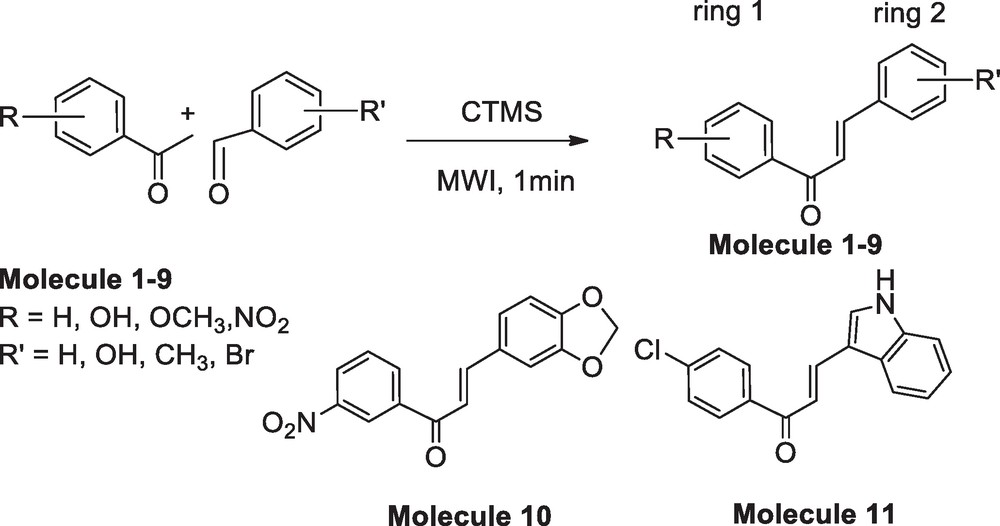
The reaction scheme for the synthesis of 1,3-diphenyl-2-propenone derivatives in 1 min.
A set of five bacterial test pathogens namely Escherichia coli, Staphylococcus aureus, Bacillus subtilis, Proteus vulgaris and Pseudomonas aeruginosa was selected for their antibacterial activity. Another set of fungus pathogens namely Aspergillus niger, Fusarium oxysporum, F. oxysporum ciceri and Candida albicans was considered to react with the synthesized chalcones to evaluate antifungal behavior. We also examined the microbial activity of acetate derivative of molecule 3 (3-Ac) to find the possibility of extending the work to other derivatives.
2 Results and discussion
2.1 Synthesis of chalcones
The use of chlorotrimethylsilane (CTMS) [20] in organic reactions either as a source of the trimethylsilyl group or as an anhydrous source of chlorides has been demonstrated over the years. Functional groups such as alcohols and amines readily undergo reaction with trimethylsilyl chloride, giving trimethylsilyl ethers and trimethylsilyl amines. Several articles have been published by this research group in order to establish the role of CTMS:
- • in the deoxygenation reaction [21,22] ;
- • and further deoxygenation via stereospecific fashion [23] of vicinal-diols;
- • cleavage of ethers [24] etc.
Transformation of amides into esters by the use of CTMS was reported elsewhere [25]. In continuation to explore prominent status, we now introduce CTMS as catalyst in the synthesis of chalcone. Depending on the reactivity of carbonyl group, an aldehyde or ketone adds to the carbonyl group of another aldehyde or ketone to give a β-hydroxyaldehyde or β-hydroxyketone as an intermediate of the crossed-aldol reaction, which can lead to a number of different products. Chalcones are generally synthesized through the Claisen-Schmidth condensation (Scheme 2) and they are stable products because of extended conjugation. Here, we present the synthesis of chalcone using CTMS via a greener and solvent-free environment. Environmentally friendly solvents, water and ethanol, were used during work-up and recrystallization. The catalysts can easily be removed by water from the reaction mixture. The results are presented in Table 1. The trans-geometry of the chalcones was ascertained by the high coupling constant of the vinylenic hydrogens (J ≈ 15–16 Hz) in the 1H NMR spectra.
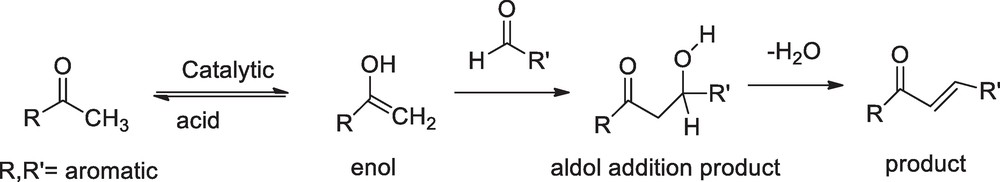
The proposed mechanism of 1,3-diphenyl-2-propenone derivative synthesis consist of the formation of aldol addition adduct followed by acid catalyzed dehydration.
Synthesis of chalcones in presence of chlorotrimethylsilane under microwave irradiation.
| Entry | Chalcone | Time (min) | Yield (%) | Ref. |
| 1 | 1 | 91 | [26] | |
| 2 | 1 | 89 | [26] | |
| 3 | 1 | 90 | [26] | |
| 4 | 1 | 88 | [27] | |
| 5 | 1.5 | 86 | [26] | |
| 6 | 1 | 94 | – | |
| 7 | 1 | 93 | – | |
| 8 | 1 | 91 | – | |
| 9 | 1 | 89 | – | |
| 10 | 1 | 96 | [28] | |
| 11 | 1.5 | 78 | [29] |
To establish the generality of a more efficient and faster synthetic route for the synthesis of chalcone, we considered substrates with different functional groups like –OH, –NO2, –OCH3, –Br, –Cl, etc. and optimized the reaction condition at ≤ 1 min with 85 to 96% yield. Molecules 6, 7, 8, 9 were detected as new entries to the chalcone family. However, the synthesis of 1–5 and 10 were reported in the literature via solution based chemistry. Heterocyclic rings such as piperinol, indole etc. are common in biological molecules. Therefore, we have synthesized chalcones 10 and 11 introducing piperinol and indole moiety under this optimized condition.
The electron withdrawing group in ring 1 enhances the eletropositivity on the carbonyl carbon of the aldehyde that makes the nucleophilic addition more effective by the enolate. This phenomenon reflects in high-yield and low reaction times for molecules 6, 7 and 10. Extended conjugation by indole ring in molecule 11 stabilizes the enolate; therefore the nucleophile is less prone to the addition reaction and results in moderate yield (78%). Presence of good hydrogen bonding groups like OH, NO2, etc. indicates higher stability suggesting high melting points.
2.2 Single crystal X-ray diffraction study
Study of structure activity relationship by determining crystal structure has become a mainline interdisciplinary subject today that deals with the self-assembly of molecules to develop protocols for predicting and controlling the structure and their functional properties. Single crystal structures of molecules 1, 2, 4, 5 were reported [30]. We determined single crystal X-ray structure for molecules 6, 7, 8 and 10 (ORTEP displayed in Fig. 1). Crystallographic data for these structures are summarized in Table 2. Bad quality and fine needles for molecule 8 did not show better quality diffraction indicating high R1 and wR2 values. Molecular assemblies by hydrogen bond and other weak interactions are presented in the supporting information that showed how the intermolecular interactions like OH···O and CH···O hydrogen bonds arranged in their molecular packing in the crystals (Supp. Info. Figure S2). No solvent molecules were found in the crystal lattice. The follow-up microbial activity study of this set of molecules illustrate how functional groups like CH3O (methoxy), OH (hydroxyl), halogens influence the activity levels for the selected pathogens.
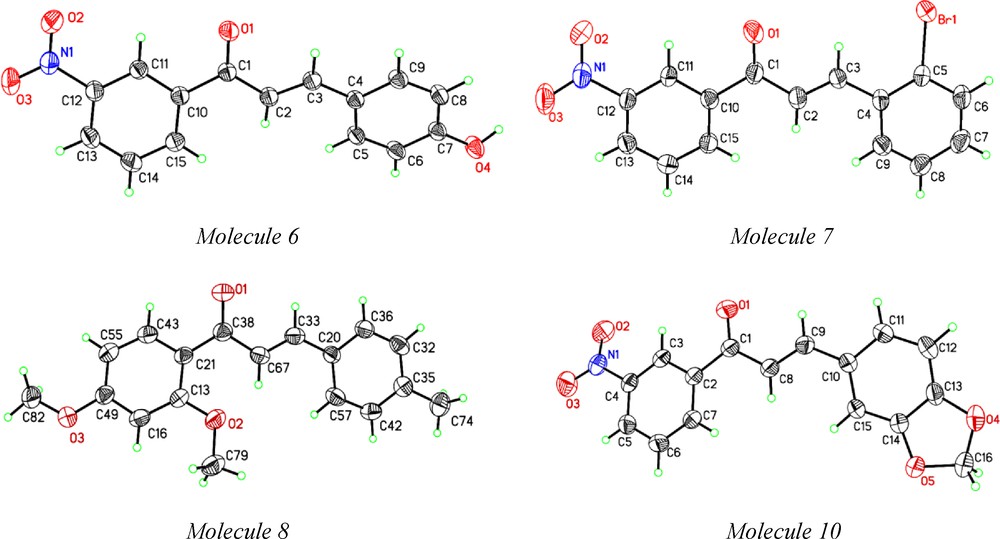
ORTEP of molecules 6, 7, 8 and 10 at 35% probability ellipsoid.
Crystallographic data of molecules 6, 7, 8 and 10. CIFs are provided as supplementary information.
| Molecule 6 | Molecule 7 | Molecule 8 | Molecule 10 | |
| Empirical formula | C15H11NO4 | C15H10BrNO3 | C18H18O3 | C16H11NO5 |
| Formula weight | 269.25 | 332.15 | 282.32 | 297.26 |
| Crystal system | Monoclinic | Monoclinic | Orthorhombic | Orthorhombic |
| Space group | P 21/c | P 21/n | Pbca | P212121 |
| a (Å) | 8.1320(16) | 7.4770(15) | 11.591(2) | 7.369(2) |
| b (Å) | 15.101(3) | 12.816(3) | 7.5870(15) | 11.313(4) |
| c (Å) | 10.474(2) | 14.213(3) | 34.411(7) | 16.228(5) |
| α (deg) | 90 | 90 | 90 | 90 |
| β (deg) | 95.09 (3) | 94.98(3) | 90 | 90 |
| γ (deg) | 90 | 90 | 90 | 90 |
| V (Å3) | 1281.2 (4) | 1356.8(5) | 3026.1(10) | 1352.9(7) |
| Dcalcd (g cm−3) | 1.396 | 1.626 | 1.239 | 1.459 |
| μ (mm−1) | 0.103 | 3.035 | 0.083 | 0.110 |
| θ range | 2.37–29.61 | 2.14–25.00 | 1.18–25.50 | 2.51–29.63 |
| Z | 4 | 4 | 8 | 4 |
| R1 [I > 2 σ(I)] | 0.0608 | 0.0728 | 0.1419 | 5.33 |
| wR2 (all) | 0.1624 | 0.1948 | 0.3394 | 12.33 |
| Goodness-of-fit | 1.123 | 1.252 | 1.387 | 1.144 |
| T (K) | 298 | 298 | 298 | 298 |
2.3 Antimicrobial activity
The investigation of the relationship between molecular structure, functionality and biological activity is the most effective way to design potential drugs. Using data from microbial activity, cytotoxicity studies, etc. one can ascertain the structural attributes responsible for the activity. For example, the presence of a 3′, 4′, 5′-trimethoxy substitution on the acetophenone ring is beneficial to biological activity [4]. Thus, with increase in number of methoxy groups, the cytotoxicity increases. However, the presence of a nitro group generally resulted in a poor activity. Both minimum inhibitory concentration (MIC) and minimum fungicidal concentration (MFC) were determined [31] in the culture tube containing 4.5 mL of either nutrient broth or M-33 broth or molten M2 agar. Final volume was adjusted to 5 mL after addition of culture media and the required antibiotic solution. Spore suspensions were prepared in Ringer's solution and adjusted to a final inoculums size of 3 × 105 colony forming units (CFU)/mL. After inoculation, the culture tubes were shaken well and then incubated at 35 °C for 24 h for pathogenic bacteria, 48 h for fungi. Incubated broth showed visible turbidity in both control and the tubes with sub-lethal doses of antibiotic.
MICs of bacterial test pathogens were determined and summarized in Tables 3 and 4, after a definite period of incubation by removing 10 mL of the contents from each tube showing no visible growth and spreading them onto agar plates of specific medium (Fig. 2). The plates were incubated at 35 °C and growth was carefully examined under microscope. MFCs were determined in potato dextrose agar medium following the same procedure and incubated for 72 h. MFC of an antibiotic was defined as the lowest concentration at which 95% of the inoculums were killed. MICs of the bioactive molecules were defined as the lowest concentration at which there was 100% inhibition of growth compared to antibiotic free control.
Antimicrobial activity measured for selected pathogen as inhibition zone in mm scale.
| Molecule | Antibacterial activity (inhibition zone in mm) | Antifungal activity (inhibition zone in mm) | |||||||
| Staphylococcus aureus | Bacillus subtilis | Escherichia coli | Pseudomonas aeruginosa | Proteus vulgaris | Candida albicans | Aspergillus niger | Fusarium oxysporum | F. oxysporum ciceri | |
| 1 | 13 | 11 | – | – | – | 13 | – | 14 | 15 |
| 2 | < 10 | 11 | – | 10 | – | 13 | – | – | – |
| 3 | 19 | 22 | 11 | – | – | – | – | 10 | – |
| 4 | – | – | 10 | – | – | – | – | – | – |
| 5 | – | 12 | – | – | – | – | – | – | – |
| 6 | – | – | – | – | – | – | – | – | – |
| 7 | – | – | – | – | – | – | – | 11 | – |
| 8 | – | – | – | – | – | – | – | – | – |
| 9 | – | – | – | – | – | 10 | – | – | – |
| 10 | – | – | – | < 10 | – | – | – | – | – |
| 11 | – | < 10 | – | – | – | 10 | – | – | – |
| 3-Ac | 17 | 15 | – | – | – | – | – | < 10 | – |
| Control | 17 | 17 | 17 | 17 | 17 | 19 | 19 | 19 | 19 |
Minimum inhibitory concentration and minimum fungicidal concentration of the active compounds.
| Molecule | Minimum inhibitory concentration (MIC, μg/mL) | Minimum fungicidal concentration (MFC, μg/mL) | |||||
| Staphylococcus aureus | Bacillus subtilis | Pseudomonas aeruginosa | Escherichia coli | Candida albicans | Fusarium oxysporum | F. oxysporum ciceri | |
| 1 | 150 | 200 | – | – | 150 | 150 | 100 |
| 2 | 150 | 100 | 200 | – | 200 | – | – |
| 3 | 100 | 50 | – | 150 | – | 200 | – |
| 4 | – | – | – | 150 | – | – | – |
| 5 | – | 100 | – | – | – | – | – |
| 6 | – | – | – | – | – | – | – |
| 7 | – | – | – | – | – | 150 | – |
| 8 | – | – | – | – | – | – | – |
| 9 | – | – | – | – | 250 | – | – |
| 10 | – | – | 250 | – | – | – | – |
| 11 | – | 150 | – | – | 250 | – | – |
| 3-Ac | 100 | 150 | – | – | – | 300 | – |
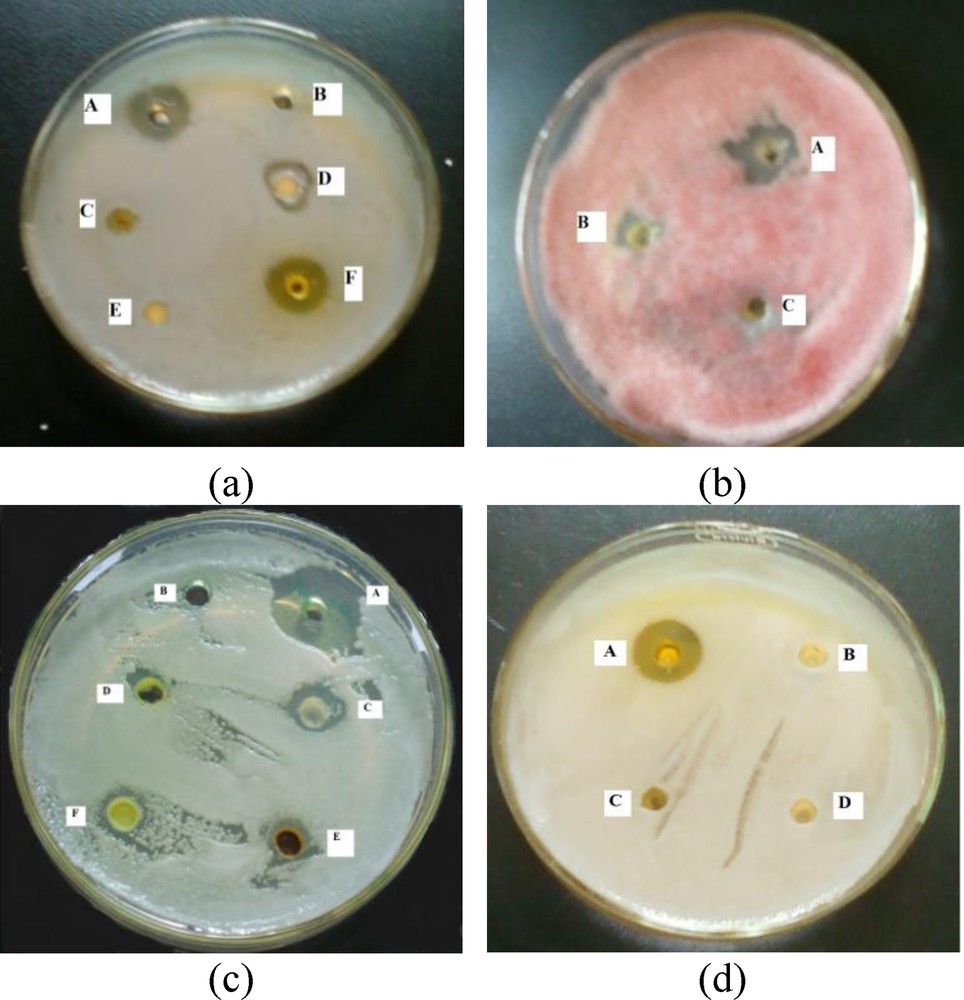
(a) Snapshots from the microbial activity experiments by synthesized chalcones on bacterial and fungal pathogens. (a) Activity against Staphylococcus aureus [A = positive control with neomycin sulphate, B = negative control with DMSO, C = molecule 2, D = molecule 1, E = molecule 4, F = molecule 3]; (b) Activity against F. oxysporum ciceri [A = molecule 1, B = molecule 3, C = molecule 2]; (c) Activity against Candida albicans [A = positive control with amphotericin B, B = negative control with DMSO, C = molecule 1, D = molecule 3, E = molecule 11, F = molecule 9]; (d) Activity against S. aureus [A = molecule 3-Ac, B = molecule 7, C = molecule 8, D = molecule 6]. Masquer
(a) Snapshots from the microbial activity experiments by synthesized chalcones on bacterial and fungal pathogens. (a) Activity against Staphylococcus aureus [A = positive control with neomycin sulphate, B = negative control with DMSO, C = molecule 2, D = molecule 1, E = molecule 4, F = molecule 3]; (b) Activity ... Lire la suite
Molecules 1, 2 and 3 exhibited both antibacterial and antifungal activities in preliminary screening (Fig. 2). However, 9 and 11 showed antifungal activity against C. albicans, a nosocomial human pathogen. The molecules 4, 10 and 11 showed very weak antibacterial activity. The acetate of molecule 3 (3-Ac) showed good antibacterial activities against S. aureus and B. subtilis. It showed less activity against F. oxysporum, a plant pathogen while, molecule 7 showed good activity against this plant pathogen. Molecules 6 and 8 have no activity against bacterial and fungal test pathogens and have not been considered for MIC and MFC determination.
MIC of all the compounds except molecule 10 against bacterial test pathogens was recorded in the range of 100 to 200 μg/mL. The MICs of molecules 3 and 3-Ac were recorded in the range of 50 to 100 and 100 to 150 respectively against Gram positive bacilli group of pathogens. However, the molecules 2 and 10 showing antibacterial activity against P. aeruginosa, a dreaded nosocomial pathogen, are to be noted and need further study to determine their efficacy against drug resistant P. aeruginosa. This pathogen is a frequent cause of nosocomial infections such as pneumonia, urinary tract infections (UTIs), and bacteremia. P. aeruginosa is the fourth most commonly isolated nosocomial pathogen, accounting for 10.1% of all hospital-acquired infections. MIC of molecule 1 was recorded in the range of 100 to 150 μg/mL and molecules 2 and 3 exhibited antifungal activity at 200 μg/mL.
Molecules 7 and 3-Ac have antifungal activity at 150 and 300 μg/mL, respectively. Also, the molecules 9 and 11 showed antifungal activity at 250 μg/mL scale. The products 1, 2, 9 and 11 exhibit antifungal activity against human pathogen C. albicans which causes Candidiasis: an infection of mouth, vagina, skin, stomach, and urinary tract.
About 75% of women get a vaginal yeast infection during their lifetime, and 90% of all people with HIV/AIDS develop Candida infections. Further study is needed to strengthen the efficacy of these molecules e.g. 1, 2, 9, 10 and 11 for developing antifungal drug/drug intermediates against dreaded human pathogens C. albicans and P. aeruginosa. Molecule 3 is also important for further study against Gram positive bacilli group. This study provides potential information to analyze how molecular structure and various functional groups may be correlated to their biological activities.
3 Conclusion
A very fast and efficient synthetic route has been developed for the synthesis of chalcones using trimethyl silyl chloride under microwave irradiation. Four new chalcones were synthesized and characterized by spectroscopy, thermal, and X-ray crystallography. Further study on microbial activities of these molecules revealed that molecules 1, 2, 3 and 11 show an antibacterial property, whereas molecules 1, 2, 7 and 11 are found to exhibit a promising antifungal property. We also observed that acetylation of chalcone 3 results in decrease of antibacterial activity.
4 Experimental
4.1 General remarks
Chemicals were purchased from Sigma-Aldrich and Merck and used as supplied. Microwave reactions were carried out in a Synthon 3000 (Anton Paar) microwave reactor. The multimode microwave has a twin magnetron (2.45 GHz) with maximum output power 1400 W. The melting points of the chalcones synthesized are determined on a Buchi Melting point apparatus B-540 and are uncorrected. All UV–vis spectra were obtained at Analytikjena Specord 200 UV-Vis spectrometer with a solution of 20 mM concentration in methanol. The existence of the CCCO bond in the chalcone moiety was monitored [35] using UV-visible spectroscopy. Extended conjugation throughout the whole molecule causes a bathochromic or red shift in UV–vis spectra. Molecule 1 shows primary absorption band at λ = 309.3 nm and a list of absorption bands of rest of the 10 molecules as a function of substituent and/or functional group are presented in Table S1 (supporting information). FT-IR spectra (in KBr matrix) were recorded in Perkin Elmer 2000 FT-IR spectrometer. NMR spectra were recorded at Bruker DPX 300 MHz (SFproton = 300 MHz, SFcarbon13 = 75.3 MHz) for all compounds (Fig. S1). Mass spectra were recorded on ESQUIRE 3000 Mass spectrometer. Elemental analysis was done in Perkin Elmer 2400 series instrument.
Single crystal X-ray diffraction studies were carried out for molecules 6, 7, 8 and 10. Reflections were collected on a Rigaku Saturn-724 CCD area detector with graphite monochromator Mo-Kα (λ = 0.71075 Å) radiation. Data reduction and solution were done using Crystal Clear software [32] using direct method [33] with expanded Fourier Techniques [34]. The data were collected for Lorentz and polarization effects. Structure refinement with anisotropic displacement parameters for non-H atoms was performed. Hydrogen atoms on O and N atoms were located from difference electron density maps in all crystal structures. All CH atoms were fixed geometrically. A check of the final CIF file did not show any serious alerts. Packing diagrams (Supp Info) were prepared in Mercury 2.3 version.
4.2 General procedure for the synthesis of chalcones
Chalcones were prepared by mixing acetophenone (500 mg, 4.16 mmol) and benzaldehyde (441 mg, 4.16 mmol) or their derivatives in equivalent molar ratio with 0.2 equivalent of chlorotrimethylsilane (CTMS) as catalyst in a mortar and pestle and then the mixture was placed under microwave irradiation for approximately 1 min. The formation of product is very fast, and can be visually monitored by a color change to yellow or brown. The product is then washed with water and recrystallized from ethanol.
4.3 Spectral data of compounds
4.3.1 1,3-Diphenylpropenone (1)
Yield 789 mg (91%); mp 56 °C, Lit.26 (56–57 °C); m/z 208; IR (KBr, cm−1) 1662, 1604, 1575, 1494, 1448, 1336, 1305, 1215, 1176, 1016, 746, 688; 1H NMR (CDCl3) δ 7.81 (1H, d, J = 15 Hz), 8.02 (2H, d, J = 6 Hz), 7.40–7.86 (9H, overlapping m, ArH); Anal. Calcd for C15H12O: C 86.51; H 5.81. Found: C 86.34; H 4.98.
4.3.2 3-(4-Hydroxyphenyl)-1-phenylpropenone (2)
Yield 831 mg (89%); mp 182 °C, Lit.26 (180–181 °C); m/z 224; IR (KBr, cm−1) 3244, 1650, 1580, 1562, 1512, 1350, 1284, 1218, 1168, 1041, 979, 834, 725; 1H NMR (CDCl3) δ 5.76 (1H, s, OH), 6.91 (2H, d, J = 9 Hz), 7.38–7.58 (6H, m), 7.78 (1H, d, J = 15 Hz), 8.01 (2H, d, J = 6.0 Hz); Anal. Calcd for C15H12O2: C 80.34; H 5.39. Found: C 80.65; H 4.46.
4.3.3 1,3-Bis-(4-hydroxyphenyl)propenone (3)
Yield 794 mg (90%); mp 198 °C, Lit.26 (196–198 °C); m/z 240; IR (KBr, cm−1) 3778, 3770, 2378, 2310, 1726, 1639, 1589, 1438, 1338, 1269, 1215, 1161, 1031, 983, 817; 1H NMR (CD3OD) δ 6.84–6.92 (4H, m), 7.56–7.63 (3H, m), 7.75 (1H, d, J = 15 Hz), 8.01 (2H, d, J = 9 Hz); Anal. Calcd for C15H12O3: C 74.99; H 5.03. Found: C 73.86; H 4.79.
4.3.4 3-(4-Methoxyphenyl)-1-phenylpropenone (4)
Yield 873 mg (88%); mp 75 °C, Lit.27 (75 °C); m/z 238; IR (KBr, cm−1) 3435, 1658, 1601, 1577,1512, 1445, 1340, 1264, 1214, 1171, 1018, 985, 825, 779, 689; 1H NMR (CDCl3): δ 3.86 (3H, s, OCH3), 6.95 (2H, d, J = 9 Hz), 7.39–7.62 (6H, m), 7.82 (1H, d, J = 15 Hz), 8.01 (2H, d, J = 9 Hz); Anal. Calcd for C16H14O2: C 80.65; H 5.92. Found: C 80.23; H 4.65.
4.3.5 1,3-Bis(4-Methoxyphenyl)propenone (5)
Yield 768 mg (86%); mp 100 °C, Lit.26 (100–102 °C); m/z 268; IR (KBr, cm−1) 1653, 1595, 1570, 1510, 1421, 1340, 1292, 1255, 1217, 1170, 1112, 1026, 977, 821, 607; 1H NMR (CDCl3) δ 3.86 (3H, s, OCH3), 3.89 (3H, s, OCH3) 6.92–7.00 (4H, m), 7.43 (1H, d, J = 15 Hz), 7.61 (2H, d, J = 9 Hz), 7.78 (1H, d, J = 15 Hz), 8.05 (2H, d, J = 9 Hz); Anal. Calcd for C17H16O3: C 76.10; H 6.01. Found: C 76.44; H 5.54.
4.3.6 3-(4-Hydroxyphenyl)-1-(3-nitrophenyl)propenone (6)
Yield 766 mg (94%); mp 196 °C; m/z 269; IR (KBr, cm−1), 3392, 1736, 1652, 1581, 1558, 1514, 1457, 1377, 1346, 1236, 1217, 1173, 1091, 985, 839; 1H NMR (DMSO-d6) δ 6.85 (2H, d, J = 9 Hz), 7.74–7.89 (5H, m), 8.47 (1H, d, J = 6 Hz), 8.57 (1H, d, J = 9 Hz), 8.8 (1H, s); 13C NMR (DMSO-d6) 116.3, 118.1, 123.0, 126.0, 127.5, 130.9, 131.9, 134.9, 139.5, 146.6, 148.5, 161.0, 187.7(CO); Anal. Calcd for C15H11NO4: C 66.91; H 4.12; N 5.20. Found: C 66.68; H 3.96; N 4.89.
4.3.7 3-(2-Bromophenyl)-1-(3-nitrophenyl)propenone (7)
Yield 935 mg (93%); mp 151 °C; m/z 331 & 333; IR (KBr, cm−1) 1668, 1613, 1598, 1530, 1465, 1437, 1349, 1267, 1213, 1086, 1026, 976; 1H NMR (CDCl3) δ 7.33 (1H, t, J = 9 Hz), 7.38–7.46 (2H, m), 7.68 (1H, d, J = 9 Hz), 7.71–7.79 (2H, m), 8.24 (1H, d, J = 12 Hz), 8.37 (1H, d, J = 6 Hz), 8.47 (1H, d, J = 6 Hz), 8.84 (1H, s), 13C NMR (CDCl3) 123.4, 123.6, 126.2, 127.2, 127.8, 128.0, 130.0, 131.9, 133.7, 134.2, 134.4, 139.1, 145.1, 148.3, 188.0(CO); Anal. Calcd for C15H10BrNO3: C 54.24; H 3.03; N 4.22. Found: C 55.54; H 3.34; N 4.52.
4.3.8 1-(2,4-Dimethoxyphenyl)-3-p-tolylpropenone (8)
Yield 713 mg (91%); mp 111 °C; m/z 282; IR (KBr, cm−1) 1649, 1606, 1500, 1463, 1417, 1327, 1280, 1251, 1211, 1163, 1128, 1024, 981, 800, 744; 1H NMR (CDCl3) δ 2.38 (3H, s, CH3), 3.87 (3H, s, OCH3), 3.90 (3H, s, OCH3), 6.46 (1H, s), 6.57 (1H, d, J = 9 Hz), 7.21 (2H, d, J = 9 Hz), 7.43–7.51 (3H, m), 7.68 (1H, d, J = 15 Hz), 7.80 (1H, d, J = 12 Hz); 13C NMR (CDCl3) 21.5 (CH3), 55.5 (OCH3), 55.7 (OCH3), 98.6, 105.1, 122.3, 126.2, 128.3, 129.6, 132.6, 132.8, 140.4, 142.2, 160.3, 164.1, 190.7(CO); Anal. Calcd for C18H18O3: C 76.57; H 6.43. Found: C 75.82; H 6.36.
4.3.9 1-(2,4-Dimethoxyphenyl)-3-(4-hydroxy-3-methoxyphenyl)propenone (9)
Yield 776 mg (89%); sticky-solid; m/z 314; IR (KBr, cm−1) 3381, 1643, 1600, 1573, 1514, 1504, 1462, 1454, 1427, 1377, 1328, 1269, 1211, 1161, 1126, 1028, 979, 823, 754; 1H NMR (CDCl3) δ 3.82 (9H, s, OCH3), 6.45 (1H, s), 6.51 (1H, d, J = 6 Hz), 6.94 (1H, d, J = 9 Hz), 7.04 (1H, s), 7.12 (1H, d, J = 6 Hz), 7.39 (1H, d, J = 15 Hz), 7.64 (1H, d, J = 15 Hz), 7.74 (1H, d, J = 9 Hz); 13C NMR (CDCl3) 55.4 (OCH3), 55.6 (OCH3), 55.7 (OCH3), 98.4, 105.2, 110.5, 115.2, 121.9, 122.8, 124.4, 127.5, 132.6, 143.1, 147.2, 148.4, 160.3, 164.1, 190.9(CO).
4.3.10 3-Benzo[1,3]dioxol-5-yl-1-(3-nitrophenyl)propenone (10)
Yield 864 mg (96%); mp 149 °C, Lit.28 (149 °C); m/z 297; IR (KBr, cm−1) 1661, 1612, 1588, 1528, 1447, 1348, 1248, 1037, 930, 806; 1H NMR (CDCl3) δ 6.06 (2H, s, CH2), 6.89 (1H, d, J = 9 Hz), 7.16 (1H, s), 7.21(1H, d, J = 6 Hz), 7.40 (1H, d, J = 15 Hz), 7.75 (1H, t, J = 9 Hz), 7.85 (1H, d, J = 15 Hz), 8.36 (1H, d, J = 6 Hz), 8.46 (1H, d, J = 9 Hz), 8.83 (1H, s); 13C NMR (CDCl3) 101.8 (CH2), 106.7, 108.8, 118.5, 123.1, 125.9, 126.9, 128.8, 129.8, 134.0, 139.7, 146.6, 148.3, 148.5, 150.5, 187.8(CO); Anal. Calcd for C16H11NO5: C 64.65; H 3.73; N 4.71. Found: C 64.21; H 3.37; N 3.81.
4.3.11 1-(4-Chlorophenyl)-3-(1H-indol-3-yl)propenone (11)
Yield 711 mg (78%); mp 57 °C, Lit.29 (57 °C); m/z 281; IR (KBr, cm−1) 3263, 1641, 1589, 1573, 1550, 1427, 1346, 1273, 1246, 1174, 1134, 1089, 1039, 1010, 974, 812, 740; 1H NMR (CDCl3) δ 7.31–7.52 (7H, m), 7.62 (1H, d, J = 15 Hz), 8.02 (2H, d, J = 9 Hz), 8.14 (1H, d, J = 15 Hz), 8.68 (1H, s, NH); Anal. Calcd for C17H12ClNO: C 72.47; H 4.29; N 4.97. Found: C 72.88; H 4.65; N 3.98.
4.4 Antimicrobial activity study
The antibacterial activity in the preliminary screening were carried out in nutrient agar (NA) medium (purchased from Hi-Media Ltd., Mumbai) using Gram positive test organisms, S. aureas (MTCC 96), B. subtilis (MTCC 441), P. vulgaris, and Gram negative bacteria, Echerichia Coli (MTCC 739), P. aeroginosa. Antifungal assays were carried out in potato dextrose agar (PDA) using fungal test pathogens; A. niger, F. oxysporum (NCIM 1281), F. oxysporum ciceri and C. albicans (MTCC 227).
In-vitro assay for antimicrobial properties were carried out against bacterial and fungal test pathogens using agar well diffusion method [36]. Bacterial and fungal suspensions of approximately 1.5 × 108 cells or spores/mL in sterile normal saline water were prepared from fresh cultures [37]. A amount of 50 μl from these suspensions was seeded uniformly by using a glass spreader over NA plates for bacteria and PDA for fungi. Wells of 6 mm diameter were made on these culture plates using a sterile cork borer. An amount of 10 mg of each pure compound from each sample was dissolved in 1 mL DMSO. From these stock solutions, 100 μL was loaded into each well. The bacterial plates were incubated at 37 °C for 24 h while fungal plates were incubated at 25 °C for 3 days. Antimicrobial activity was determined by measuring the diameter of inhibition zones (mm) of test microorganisms around the well after incubation. Neomycin sulphate (30 μg) and amphotericin B (50 μg) were used as positive control while dimethyl sulphoxide was taken as negative control.
Acknowledgement
We thank the director, CSIR-NEIST, Jorhat for extending research facilities during the work.
Vous devez vous connecter pour continuer.
S'authentifier