

t-Butanesulfinamide 1 (Fig. 1) has found a huge field of applications for the synthesis of optically pure amines via the intermediary of t-butanesulfinyl imines 2, either by nucleophilic addition or by electrophilic trapping of the sulfinylenamine after deprotonation by a base [1,2].
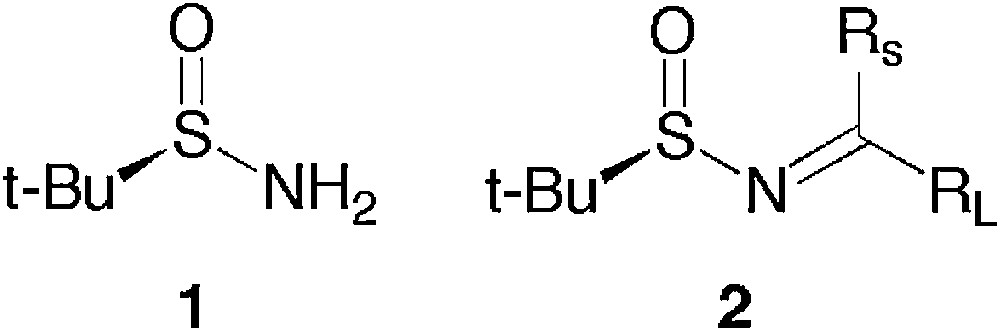
t-Butanesulfinamide and t-butanesulfinyl imines.
The use of this chiral auxiliary is highly wide in scope via the sulfinylimines and it has also been extended to higher oxidation levels as an elegant method to access chiral amide or esters by alkylation of N-sulfinyl imidates 3 (Scheme 1) [3].
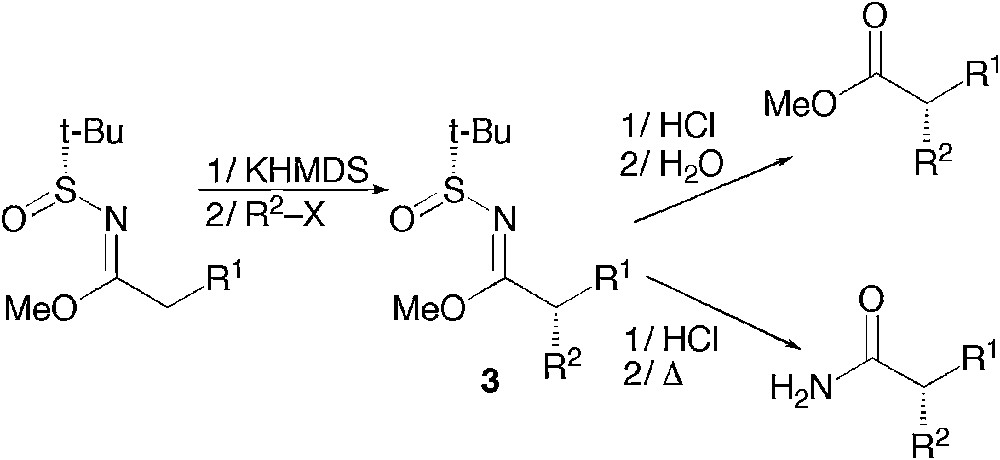
Asymmetric synthesis of carboxylic derivatives by alkylation of N-sulfinyl imidates.
Moreover, whereas removal and/or recovery of the chiral auxiliary remains one general concern in these diastereoselection-based asymmetric synthetic methods, the t-butane sulfinamides obtained easily release their sulfinyl moiety via a simple anhydrous acidic treatment, which also allows the isolation of the ammonium salt (and potentially the recovery of the sulfinyl chloride) [4]. t-Butanesulfinamide has recently found new applications since reports have mentioned its use in t-butanesulfinyl-based ligands to be used in transition metal asymmetric catalysis, especially rhodium-catalyzed 1,4-addition of organoboronic acids to activated double bonds (Fig. 2) [5–7]. Expectedly, it has been shown that coordination of the sulphur atom to the transition metal, such as in 4 provides a good stereochemical induction. Moreover, the sulfinyl moiety attached to nitrogen has also been recently widely exploited in the development of organocatalytic methods where it has been shown either to act as an organocatalyst, [8] or to modulate the properties of urea and thiourea either by acidifying the adjacent proton or via the H-acceptor ability of the SO group [9–14]. This illustrates the fact that the use of sufinyl adducts is far from being restricted to a role as chiral auxiliary and therefore, the establishment of methods for opening routes to new sulfinyl containing compounds is a field currently regaining interest.

Structure of a rhodium sulfinamide/alkene dimer.
In the course of our studies aiming at developing a new route to t-butanesulfinamide 1, we have been willing to explore the reactivity of t-butane sulfinylphthalimide 6 toward oxygen- and nitrogen-centred nucleophiles, as this could not only represent a desirable new route to t-butanesulfinamide 1, but also a potential access to chiral sulphur bidentate ligands. Although not widely developed and used, sulfinylphthalimides have been known for many decades [15].
Our first attempt to access t-butane sulfinylphthalimide 6 has been the most straightforward that can be identified, i.e. the condensation of potassium phthalimide on t-butanesulfinyl chloride 5 (Scheme 2). Although the yield was very good on our first attempt, we had serious concerns regarding the reproducibility of this reaction.

Synthesis of t-butanesulfinyl phthalimide from t-butanesulfinyl chloride.
This seems to be mainly due to the poor stability of t-butanesulfinyl chloride 5, which was then isolated only in low yields regardless of its synthetic path. We therefore explored an alternate approach, which could reliably allow us to access t-butane sulfinylphthalimide 6 on a multi-gram scale, in a cost-effective fashion.
The t-butane sulfinylphthalimide 6 might also be prepared in two stages from t-butane mercaptan 7 by the action of chlorine according to a known procedure in industry to yield the unstable sulfenyl chloride 8 (Scheme 3). [16] By quenching 8 in situ with a source of phthalimide, one can expect to access the sulfenylphthalimide 9, which then only requires oxidation to yield 6.
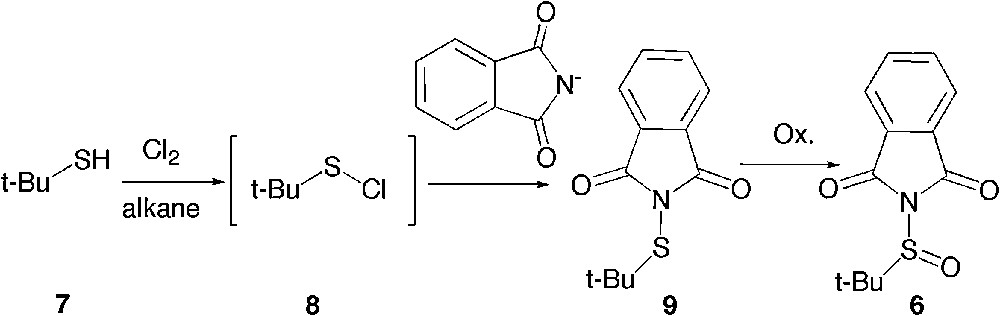
Alternate access to t-butanesulfinylphthalimide.
For the sake of practicality at the laboratory scale, and keeping in mind that the reaction would ideally be carried out on multi-gram scale, we found it advantageous to replace chlorine by a more convenient oxidant. Moreover, starting from the disulfide of t-butylmercaptan was favoured, not only for cost-effectiveness, but also because t-butyl mercaptan exhibits a significant disadvantageous smell. Yet, the selective cleavage of the S–S bond has been reported as a highly unfavoured process (Scheme 4) [17].

Selectivity issue in the oxidative cleavage of di-t-butyldisulfide.
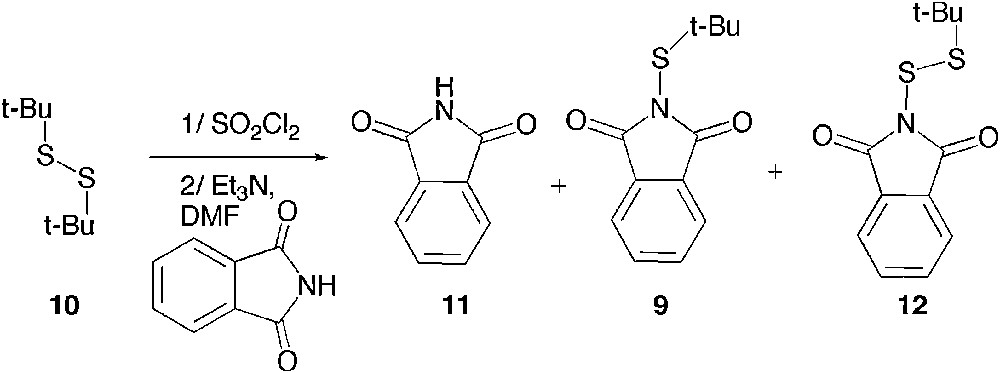
We nevertheless decided to tackle this synthetic challenge and first explored the possibility to use N-bromophthalimide, which could be both the oxidant and the source of phthalimide [18]. The reaction was ideally easy to set up as it was carried out by mixing the di-t-butyl disulfide 10 and N-bromophthalimide under refluxing chloroform (Scheme 5); yet, only a poor mass recovery was observed and the isolated solid happened to be a mixture of phthalimide 11 with a minor amount of sulfenylphthalimide 9, and a more important quantity of thiosulfenylphthalimide 12.

Reaction of 10 with bromophthalimide.
As illustrated in Scheme 4, 9 is thought to arise from the desired and expected cleavage of the S–S bond, whereas 12 is formed by the cleavage of the C–S bond favoured in the case of di-t-butyl mercaptan and occurring with a loss of isobutene. Because of the lack of efficiency and since the separation of these two compounds appeared tedious, we changed the oxidant to sulfuryl chloride (a source of Cl+ previously reported to cleave the S–S bond of a primary alkyl disulfide) [19] in order to increase both the efficiency and the 9:12 ratio (Scheme 6). We also explored the effect of the solvent and the temperature on the reaction outcome (Table 1).
Formation of t-butanesulfenyl phthalimide form 10.
Identification of the optimal reaction conditions for the synthesis of 9.
| Entry | Solvent | Temperature (°C) | Recovery of 11 | Ratio 9:12 | Yield of 9 (%) |
| 1 | None | –30 | 100 | _ | 0 |
| 2 | Pentane | –30 | 100 | _ | 0 |
| 3 | None | 0 | 100 | _ | 0 |
| 4 | Pentane | 30 | 17 | 92/8 | 76 |
| 5 | Pentane | 36 | 50 | 83/17 | 41 |
| 6 | Heptane | 30 | 20 | 9/91 | 7 |
| 7 | Heptane | 80 | 44 | 61/39 | 34 |
The reaction was followed by GC to monitor the consumption of the disulfide before the addition to a solution of phthalimide and triethylamine (2 and 2.6 equiv of both with regard to the initial disulfide, respectively) in DMF. Precipitation was then induced by dilution with water, and the ratio 9:12 was determined from 1H-NMR. At low temperature (–30 °C and 0 °C) in pentane or under solvent-free conditions, the solid that was finally isolated after precipitation from water was only phthalimide 11. As we could not isolate any other product from this reaction, we moved to other conditions. Oppositely, when the reaction was carried out at 30 °C in pentane, a good mass recovery of a mixture of products is obtained with an interesting ratio of 92/8 in favour of the sulfenylphthalimide 9. Raising the temperature to the boiling point of pentane slightly lowers both the recovery and the ratio (entry 5). Surprisingly, running the reaction in heptane at 30 °C also allows the isolation of a significant amount of material, but in this case, the ratio between 9 and 12 was inversed in favour of the thiosulfenylphthalimide 12 (entry 6). In heptane, it is only at 80 °C that a ratio in favour of 9 was observed again (entry 7) yet with a lower efficiency compared to the reaction in pentane. We have so far no evidence allowing an explanation of these experimental observations. Nevertheless, the reaction carried out in pentane at 30 °C was found to be highly reproducible up to a 0.1 mol scale. With the 92/8 ratio between 9 and 12 at the end of the reaction, we found that the sulfenylphthalimide 9 could be isolated in 76% yield by a simple precipitation in five volumes of absolute ethanol. One interesting feature of this reaction is that no volatile derivative of t-butyl mercaptan is released and therefore no unpleasant smell was detected at any time. This is obviously the consequence of the efficient transformation of one disulfide into two sulfenylchloride.
Having an efficient and scaled-up approach to 9, we performed its oxidation to 6 under classical conditions. 6 has been quantitatively formed in the presence of a stoichiometric amount of mCPBA, as previously reported [20], or peracetic acid (Scheme 7). The latter oxidising reagent proved superior for preparative purposes as it allowed the isolation of the sulfinyl phthalimide 6 in a highly pure form in 93% yield, the acetic acid being removed upon aqueous work-up. Oppositely, the mCPBA oxidation required further purification. We have routinely carried out this oxidation on a multi-gram scale, securing an easy and reproducible access to t-butanesulfinyl phthalimide 6.
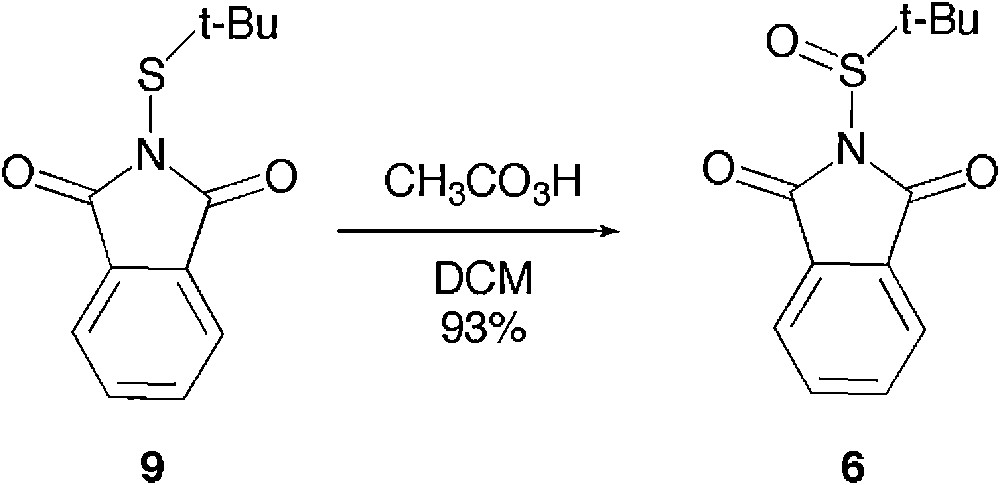
Mono-oxidation of t-butane thiophtamilide.
With this high-scale and reproducible approach, we then explored the reactivity of the t-butanesulfinyl phthalimide 6 toward oxygen- and nitrogen-centered nucleophiles.
Although a simple nucleophilic displacement of phthalimide has been reported earlier for sulfinylphthalimides 6a, upon increase of the steric hindrance of the substituent on sulphur, a complete inversion of selectivity has been achieved, driving the reaction exclusively to the ring opening of the phthalimide ring. [15] Yet, only one ring opening of 6 by a bulky secondary alkoxide leading to 13 had been described as a side reaction in this report. A similar ring opening had been previously described on a range of sulfenylphthalimides 9 with an array of nucleophiles yielding 14 [21]. Owing to this general difference of behaviour between sulfenyl- and sulfinylphthalimides, it remained unclear whether at a higher level of oxidation of the sulphur atom, one would still observe with any other nucleophiles a ring opening rather than a phthalimide substitution (Scheme 8).
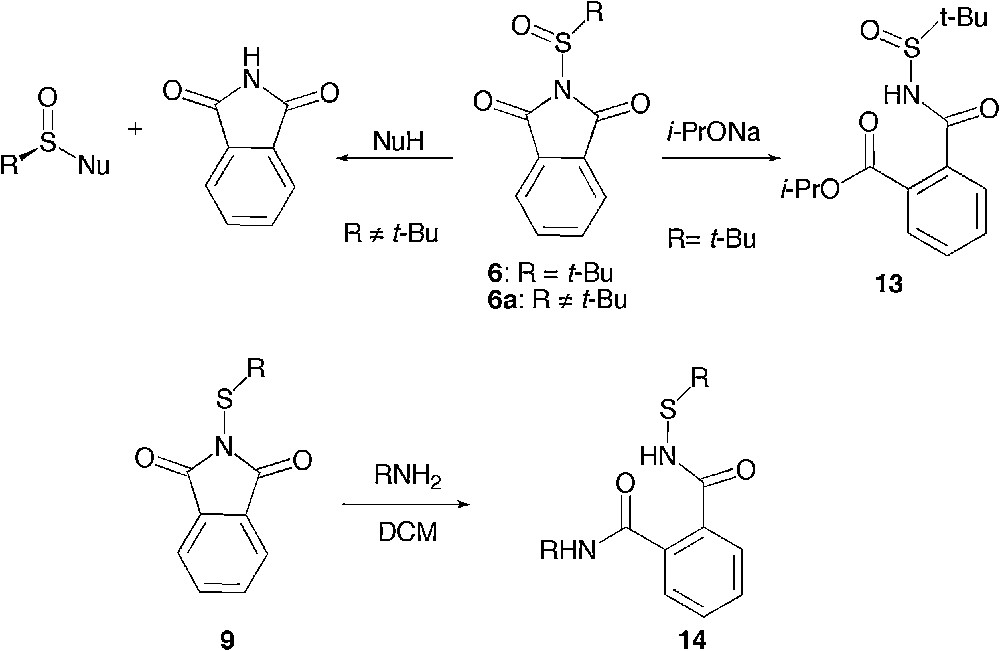
Electrophilic site and regioselectivity from the literature.
We were first wondering whether a similar reactivity would be achieved via acid catalysis. Therefore, we explored the reactivity of the t-butanesufinyl phthalimide 6 with a pro-nucleophile in the presence of a Lewis acid. We observed that although 6 is stable as a suspension in ethanol, addition of 0.5 mol% of the Lewis acidic samarium (III) triflate promotes the total consumption of the starting material in half an hour and allows the isolation, in quantitative yield, of the phthalimide ring-opened product 15, whose structure has been unambiguously determined by X-ray diffraction (Scheme 9) [22].
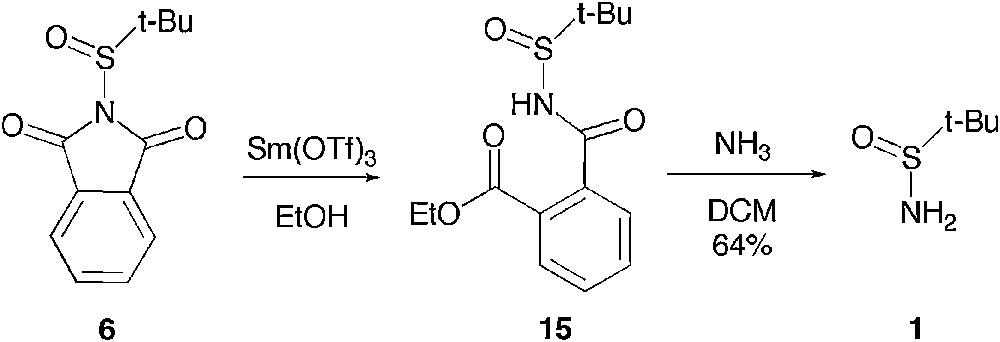
Ring opening under Lewis acid catalysis.
Interestingly, the addition of aqueous ammonia to a solution of 15 in dichloromethane allowed the isolation of t-butane sulfinamide in 64% yield. As this was establishing that a diastereo-controlled access to a derivative of 15 would provide an enantioselective access to t-butanesulfinamide 1, we tried to apply these very mild ring opening reaction conditions in the presence of a chiral alcohol such as 1-phenyl ethanol. Unfortunately, we could not expand the scope of this reaction to this bulkier alcohol, no reaction being observed even in the presence of a heavier load of catalyst. We therefore investigated the addition of amines and alkoxides to t-butanesufinyl phthalimide 6.
In order to favour π-interactions that could ease the separation of the diastereoisomers, we engaged chiral amines bearing a pendent aromatic ring in these reactions and found that addition of one equivalent of (R)-1-phenylethylamine or (S)-1-naphthylethylamine in dichloromethane allows the isolation of the desired ring-opened products 16 and 17 in 91% and 87% yields, respectively (Scheme 10). Unfortunately, although these compounds are easily and efficiently accessed, none of the techniques investigated allowed the separation of the two diastereoisomers. The addition of the sodium salts of chiral alcohols, such as (S)-1-phenylethanol or the diacetonide-d-glucose in THF has also been carried out. Yields were in these cases significantly lower (52% and 43% for 18 and 19, respectively), and again we could not perform the separation of the two diastereoisomers.

Ring opening of 6 by a range of chiral nucleophiles.
This diastereoselective approach has not allowed the desired straightforward access to optically pure t-butanesulfinamide. Nevertheless, original t-butanesulfamides have been accessed in a highly efficient fashion. They represent an original class of compounds and those (16 and 17) incorporating two H-bond donors might find applications in organocatalysis considering that the linker between these H-donors can be somehow modulated. We are currently trying to take advantage of this easy, cost efficient and scaled-up method, we have described here to generate a range of potential organocatalysts based on these original structures.
1 Experimental details
All the reactions requiring anhydrous conditions were conducted in oven-dried apparatus under an atmosphere of argon. Syringes and needles for the transfer of reagents were dried at 110 °C and allowed to cool in a desiccator over P2O5 before use. CH2Cl2, pentane, heptane were distilled from CaH2 under argon. External reaction temperatures are reported unless stated otherwise. The reactions were monitored by TLC using commercially available glass-backed plates, pre-coated with a 0.25 mm layer of silica containing a fluorescent indicator. Flash chromatography was carried out on Interchim Puriflash 430 using Interchim prepacked column (30 μ). 1H- and 13C-NMR spectra were recorded with a Bruker AV 300 or a Bruker AV 400 spectrometer. Chemical shifts are reported relative to tetramethylsilane. Coupling constants (J) are given in Hz.
1.1 t-Butane sulfenylphthalimide 9
To a solution of di-t-butyl disulfide (17.84 g, 0.1 mol) in 200 mL of pentane at 30 °C are added dropwise 9 ml (0.11 mol) of sulfuryl chloride to maintain the internal temperature of the reaction medium above 28 °C. Once the addition is completed, the reaction mixture is stirred at 30 °C for 3 h, and then slowly added to a strongly stirred solution of 26.48 g (0.18 mol) of phthalimide and 33.5 mL (0.24 mol) of triethylamine in 30 mL of DMF. After 1 h, 300 mL of water are added to the reaction mixture, yielding a white precipitate. After filtration and recrystallization from hot absolute ethanol, the title compound is obtained as a white powder (32.07 g, 76%).
1H-NMR (CDCl3, 400 MHz): 7.97 (m, 2H), 7.81 (m, 2H), 1.39 (s, 9H). 13C-NMR (CDCl3, 100 MHz): 169.1, 134.6, 132.0, 124.0, 51.1, 29.4. MS (DCI, NH3): 253 (MNH4+). IR (ATR, cm−1): 3092, 3065, 2975, 2963, 1743, 1347, 1275, 1154, 1039, 867, 796, 717, 687. HRMS (DCI, CH4): calculated for C12H13NO2S: 236.0745, found: 236.0748. MP: 131–132 °C (lit. 130–131 °C).
1.2 t-Butane thiosulfenylphthalimide 12
To a solution of di-t-butyl disulfide (17.84 g, 0.1 mol) in 200 mL of heptane at –20 °C are added 9 ml (0.11 mol) of sulfuryl chloride. After 3 h, the reaction mixture is added to a strongly stirred solution of 26.48 g (0.18 mol) of phthalimide and 33.5 mL (0.24 mol) of triethylamine in 30 mL of DMF at 20 °C. After 1 h, 600 mL of cold water are added and the resulting solid is filtrated, washed with 50 mL of water and 50 mL of pentane. Crystallisation from cold absolute ethanol yields to 14.5 g (54%) of the title compound.
1H-NMR (CDCl3, 400 MHz): 7.94 (m, 2H), 7.82 (m, 2H), 1.43 (s, 9H). 13C-NMR (CDCl3, 100 MHz): 167.6, 134.7, 132.2, 124.0, 49.3, 29.9. MS (DCI, NH3): 285 (MNH4+). IR (ATR, cm−1): 2958, 1778, 1724, 1699, 1466, 1457, 1363, 1342, 1251, 1163, 1050, 864, 792, 710. MP: 103–104 °C.
1.3 t-Butane sulfinylphthalimide 6
To a solution of 32.07 g (136.3 mmol) of t-butane thiophthalimide in 320 mL of dichloromethane at 0 °C is added dropwise a 35% solution of peracetic acid (29.62 g, 136.6 mmol) in acetic acid. After 18 h of strong stirring, 100 mL of a 5% solution of sodium thiosulfate are added to the reaction mixture. The organic phase is then washed two times with 100 mL of a 5% solution of sodium thiosulfate, dried over sodium sulphate and evaporated under reduced pressure to yield 33.86 g (93%) of the title compound as a white solid.
1H-NMR (CDCl3, 400 MHz): 7.92 (m, 2H), 7.82 (m, 2H), 1.48 (s, 9H). 13C-NMR (CDCl3, 100 MHz): 166.6, 135.0, 132.1, 124.1, 61.3, 23.5. MS (DCI, NH3): 269 (MNH4+). IR (ATR, cm−1): 3088, 3027, 2979, 1725, 1460, 1365, 1261, 1117, 1040, 864, 796, 718. HRMS (DCI, CH4): calculated for C12H13NO3S: 252.0694, found: 252.0694. MP: 136–137 °C (lit. 133–136 °C).
1.4 2-(t-butane sulfinylcarbamoyl) ethylbenzoate 15
To a suspension of t-butane sulfinylphthalimide (100 mg, 0.4 mmol) in absolute ethanol is added 23.8 mg of samarium (III) triflate (0.04 mmol). Once the solution becomes homogeneous (0.5 h), the solvent is removed under vacuum, the solid is dissolved in 20 mL of dichloromethane, washed with water and brine (2 × 10 mL), dried over sodium sulphate and concentrated under vacuum to yield the title compound (118.1 mg, 100%) as a white solid.
1H-NMR (CDCl3, 400 MHz): 8.05–7.97 (m, 1H), 7.55–7.61 (m, 4H), 4.40 (q, 2H, J = 7.2), 1.40 (t, 3H, J = 7.2), 1.33 (s, 9H). 13C-NMR (CD2Cl3, 100 MHz): 173.9, 166.3, 136.1, 132.2, 130.6, 130.0, 129.1, 127.5, 62.1, 58.1, 22.1, 14.1. MS (DCI, NH3): 315 (MNH4+). IR (ATR, cm−1): 3068, 2971, 1710, 1688, 1428, 1278, 1247, 1062, 885, 805, 703. MP: 129–131 °C.
1.5 Addition of chiral amines to t-butane sulfinylphthalimide, general procedure
To a solution of t-butane sulfinylphthalimide in 15 volumes of dichloromethane is added one equivalent of the chiral amine. The solution is heated under reflux for 16 h, concentrated to dryness and purified by flash chromatography (eluent DCM/i-PrOH: 95/5).
1.5.1 2-(2-(N-t-butane sulfinyl)N-acetyl)-N-((R)-1-phenylethyl)benzamide 16
The title product is a white solid isolated as a mixture of the two diastereoisomers (1.74 g, 91%).
1H-NMR (CD2Cl2, 400 MHz): 7.92–7.76 (m, 2H), 7.54–7.50 (m, 6H), 7.45–7.39 (m, 8H), 7.35–7.31 (m, 2H), 7.14–6.89 (m, 2H), 5.28–5.23 (m, 2H), 1.61–1.58 (m, 6H), 1.30 (s, 9H), 1.24 (s, 9H). 13C-NMR (CD2Cl2, 100,6 MHz): 143.0, 142.8, 134.9, 134.8, 132.8, 131.5, 130.7, 130.5, 130.4, 128.7, 127,9, 127,8, 127,5, 127,4, 126,2, 126,1, 56,4, 56,3, 50,1, 49,8, 22,0, 21,9, 21,5. MS (DCI): 373,0 (MH+), 390,0 (MNH4+). HRMS (DCI): calculated for C20H24N2O3S: 373,1586, found: 373,1598. IR (cm−1): 3253, 3063, 2974, 1632, 1536, 1447, 1415, 1064, 868, 763, 697.
1.5.2 2-(2-(N-t-butane sulfinyl)-N-acetyl)-N-((S)-1-naphtylethyl) benzamide 17
The title product is a white solid isolated as a mixture of the two diastereoisomers (1.85 g, 87%).
1H-NMR (CDCl3, 400 MHz): 8.17 (dd, 2 × 1H J = 8.4 Hz, J = 3.6 Hz), 7.93–7.83 (m, 2 × 3H), 7.71–7.42 (m, 2 × 6H), 7.35 (d, 2 × 1H, J = 7.2 Hz), 6.74 (d, J = 8.2 Hz), 6.62 (d, 2 × 1H, J = 8.2 Hz), 6.14–6.06 (m, 2 × 1H), 1.80 (d, 2 × 3H, J = 6.8 Hz), 1.39 (s, 9H), 1.26 (s, 9H). 13C-NMR (CDCl3, 100,6 MHz): 137.6, 137.0, 134.7, 134.6, 134.0, 133.9, 132.4, 132.0, 131.7, 131.6, 131.1, 130.9, 130.6, 129.0, 128.9, 128.8, 128.6, 127.8, 127.6, 127.3, 126.7, 126.2, 126.0, 125.4, 125.2, 123.2, 123.0, 122.9, 122.8, 56.8, 56.6, 46.0, 45.8, 22.3, 22.2, 20.8, 20.3. MS (DCI): 423.1 (MH+), 440.1 (MNH4+). HRMS (DCI): calculated for C24H26N2O3S: 423.1742, found: 423.1729. IR (cm−1 Hz): 3275, 2974, 1660, 1623, 1545, 1463, 1057, 800, 780.
1.6 Addition of chiral alcohols to t-butane sulfinylphthalimide, general procedure
To a solution of chiral alcohol in 15 volumes of distilled THF is slowly added sodium hydride (1.5 equiv of 60% suspension in mineral oil). After 1.5 h, t-butane sulfinylphthalimide (1 equiv) is added and stirring is then prolonged for 1 h. Thirty volumes of a saturated ammonium chloride solution are added dropwise. The aqueous is extracted three times with dichloromethane; the organic phases are washed with brine, dried over sodium sulphate, filtrated, and concentrated under vacuum. The residue is purified by flash chromatography (gradient elution of cyclohexane/diethyl ether: 50/50 to 0/100 Hz).
1.6.1 (S)-methylbenzyl 2-(t-butane sulfinylcarbamoyl)-1-benzoate 18
The title product is a white solid isolated as a mixture of the two diastereoisomers (0.97 g, 52%).
1H-NMR (CD2Cl2, 400 MHz): 8.03 (d, 2 × 1H, J = 7.2 Hz), 7.67–7.32 (m, 2 × 8H), 6.10 (q, 2 × 1H, J = 6.6 Hz), 1.70 (d, 2 × 3H, J = 6.6 Hz), 1.26 (s, 2 × 9H). 13C-NMR(CD2Cl2, 100,6 MHz): 141.5, 141.4, 136.4, 136.3, 132.4, 132.4, 130.3, 128.5, 128.4, 128.0, 127.9, 127.8, 126.2, 126.1, 125.8, 74.2, 57.1, 22.0, 21.9, 21.8. MS (DCI): 374 (MH+), 391 (MNH4+). IR (cm−1 Hz): 3067, 2978, 1717, 1687, 1416, 1265, 1057, 760, 736, 697.
1.6.2 Diacetone-d-glucose 2-(t-butane sulfinylcarbamoyl)-1-benzoate 19
The title product is a white solid isolated as a mixture of the two diastereoisomers (0.67 g, 43%).
1H-NMR (CD2Cl2, 400 MHz): 7.92–7.83 (m, 2 × 1H), 6.78–6.57 (m, 3 × 2H), 6.01 (d, 1H, J = 3.6 Hz), 5.98 (d, 1H, J = 3.6 Hz), 5.42–5.41 (m, 2 × 1H), 4.75 (d, 1H, J = 3.6 Hz), 4.72 (d, 1H J = 3.6 Hz), 4.36–4.27 (m, 2 × 2H), 4.14–4.01 (m, 2 × 2H), 1.55 (s, 2 × 3H), 1,42 (s, 2 × 3H), 1.35 (s, 2 × 3H), 1,31 (s, 2 × 3H), 1.29 (s, 2 × 9H). 13C-NMR (CD2Cl2, 100,6 MHz): 136.3, 136.2, 135.0, 132.6, 132.4, 132.1, 130.6, 130.5, 130.0, 128.0, 127.9, 123.9, 112.2, 112.1, 109.3, 109.2, 105.4, 105.3, 83.1, 82.9, 79.7, 79.6, 77.6, 77.5, 72.6, 72.5, 67.1, 67.0, 57.6, 57.4, 26.6, 26.5, 26.4, 26.0, 23.0, 22.0, 21.9. MS (DCI): 529 (MNH4+). HRMS (DCI): calculated for C24H33NO9S: 512,1954, found: 519,1977. IR (cm−1 Hz): 2986, 1729, 1689, 1371, 1260, 1064, 1017, 843, 736, 705.
Disclosure of interest
The authors declare that they have no conflicts of interest concerning this article.
Acknowledgements
The authors are very much thankful to Minakem holding (A.H. & G.C.), to the Conseil régional Midi-Pyrénées (G.C.) for PhD fundings, to Minakem for generous sponsorship and to the CNRS and the University of Toulouse for their support.