

1 Introduction
The steroid system has been a prime focus of research throughout scientific history and displays a variety of biological functions [1–4]. Steroidal compounds, widely used in traditional medicines, exhibit a wide range of activities, including anticancer and antibacterial [5,6], and have the potential to be developed as drugs for the treatment of a large number of diseases. They have also become the basis of many important discoveries in organic chemistry. Recently, steroidal building blocks have attracted interest with a view to modifying steroidal molecules [7,8]. Their preparation is a stimulating challenge to organic chemists, often demanding the development of new and generally useful reactions. These compounds are often non-toxic, capable of overcoming multi-drug resistance, and highly bioavailable because of their capacity of penetrating the cell wall. Several steroidal derivatives have been investigated as new curative agents for cancer, and exhibited diverse biological activities, such as antimicrobial, anti-inflammatory, anticancer ones [9–11].
The biological activity of Schiff bases containing thione (CS) was exploited in the 1960s using N-methylisatin-β-thiosemicarbazone, which was effective against smallpox and vaccinia [12,13]. 3-Aminpyridine-2-carboxaldehyde thiosemicarbazone is now being evaluated in clinical trials against several malignancies [14]. Compounds containing thione (CS) groups occupy an important position among organic reagents. Organic compounds with this structure are known to display a wide range of pharmacological activity, including anticancer, antibacterial and antifungal effects [15–18]. A good deal of research has been carried out on the potential of coordination chemistry of thiocarbohydrazones, and thiocarbohydrazide attached to carbonyl compounds having heterocyclic moiety to give potential drugs [19,20]. Further optimization of this compound is required to improve its antimicrobial activity.
Although there are many reports based on steroidal units’ modification, including derivatization, cyclization [21], there are few reports about steroidal Schiff base. Steroid is known for its abundance and importance in biological systems, which is expected to show relatively low toxicity, augmented biocompatibility [22]. Furthermore, thiourea and its related analogues have become the focus of interest in recent years on account of their pharmacological activities. According to the active principle of superposition. We hoped that thiosemicarbazone derivatives containing steroid unit and thiourea group have potent antibacterial activity. In addition, microwave synthesis is a well-established technique in green chemistry and has many advantages compared with the traditional methods, such as faster reaction times, higher yields; moreover, it is a solvent-free method [23–26]. It was being applied in many organic synthesis. Accordingly, as a further development of our green agenda [27–29], herein we report a microwave-mediated, efficient, high yield and environmentally friendly synthesis of novel steroidal bis thiocarbazone derivatives under solvent-free conditions (Scheme 1) as well as an evaluation of the antibacterial activities of these compounds against four pathogenic strains, Escherichia coli (ATCC 35218), Bacillus subtilis (ATCC 6633), Pseudomonas aeruginosa (ATCC 27853), and Staphyloccocus aureus (ATCC 6538).
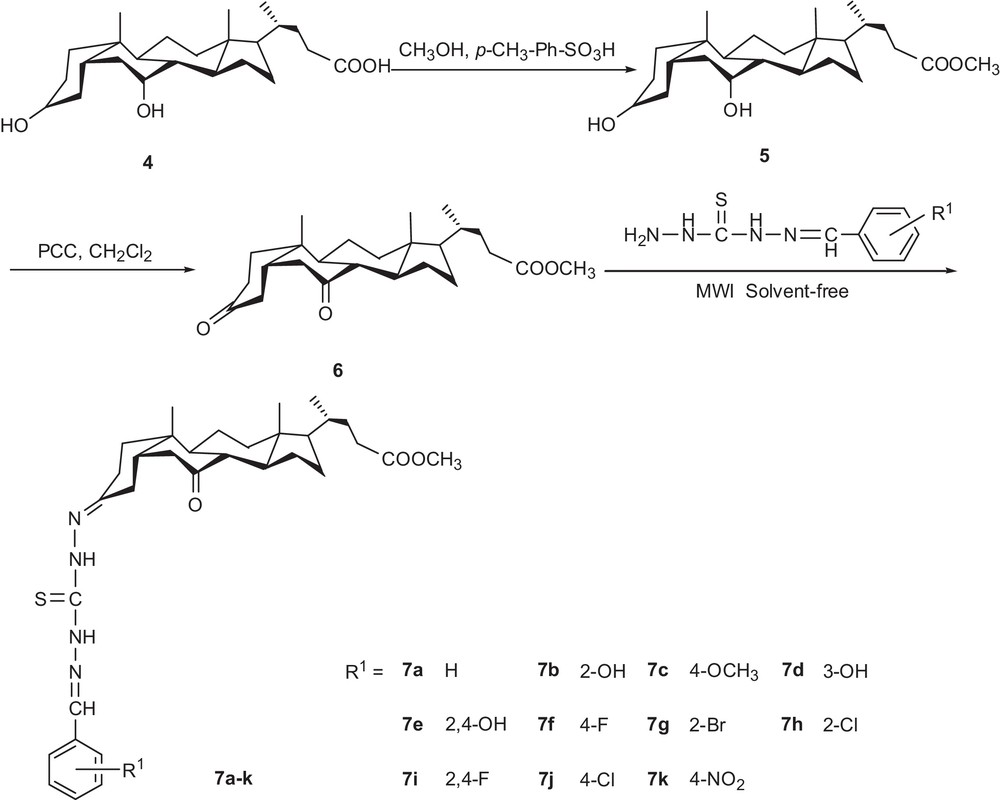
Synthetic protocol of titled compounds 7a–k.
2 Result and discussion
It is known that a carbonyl group at C-3 of steroidal scaffold is more susceptible to nucleophilic addition reactions than a carbonyl at C-7 [30]. In our quest to find optimal reaction conditions, we studied steroidal diketones with substituted benzaldehyde thiocarbohydrazones, employing conventional and microwave-mediated solvent-free thermal procedures. The conventional reaction was conducted with or without catalysts, such as HCl, acetic acid or H2SO4 by refluxing in ethanol, acetic acid, methanol, THF, DMF, or DMSO. The result showed that the yield for 7a was in the range 24–52% (Table 1). But it required a longer reaction time. So, the catalytic properties of HCl have been studied considering the synthesis of compound 7a.
Effect of the reaction conditions on the synthesis of compound 7a.
| Entry | Solvent | Catalyst | Support | Time (min) | Yield (%) |
| 1 | EtOH | – | – | 420 | 24a |
| 2 | EtOH | HCl | – | 420 | 52a |
| 3 | EtOH | acetic acid | – | 420 | 43a |
| 4 | EtOH | H2SO4 | – | 420 | 39a |
| 5 | Acetic acid | – | – | 420 | 40a |
| 6 | THF | HCl | – | 420 | 42a |
| 7 | MeOH | HCl | – | 420 | 48a |
| 8 | DMF | – | – | 420 | 32a |
| 9 | DMSO | – | – | 420 | 36a |
| 10 | – | – | Silica gel H | 5.5 | 53b |
| 11 | – | HCl | Silica gel H | 5.5 | 61b |
| 12 | – | – | Neutral Al2O3 | 5.5 | 32b |
| 13 | – | HCl | Neutral Al2O3 | 5.5 | 84b |
| 14 | – | – | K2CO3 | 5.5 | 12b |
| 15 | – | HCl | K2CO3 | 5.5 | 40b |
a Conventional method.
b Microwave method.
We then studied the effect of various supports, such as silica gel H, neutral Al2O3 and K2CO3 with or without HCl catalyst for the synthesis of compound 7a using the microwave-assisted method. Among the results obtained, the use of HCl in neutral Al2O3 received the best yield (84%) (Table 1). Finally, we evidenced a dramatic improvement in the yield and the reaction time (5.5 min) when employing a microwave-assisted method. The microwave-assisted procedure was then applied to the synthesis of compounds 7b–k.
In order to study the advantages of the microwave-promoted reaction, we compared the synthesis of compounds 7a–k using conventional heating and the microwave-assisted method under solvent-free conditions. Compared to the conventional thermal heating, the microwave irradiation technique decreased the reaction time from 420–540 min to 5.0–7.5 min and increased the yields from 31–58% to 80–89% (Table 2). Consequently, the use of the microwave technology in conjunction with solvent-free conditions allows expeditious, environmentally friendly and efficient procedures in organic methodology.
Synthesis of compounds 7a–k using microwave irradiation or conventional method.
| Compound | R1 | Conventional heating | Microwave method | TC/TMW | ||
| T (min) | Yield (%) | T (min) | Yield (%) | |||
| 7a | H | 420 | 47 | 5.5 | 84 | 76 |
| 7b | 2-OH | 480 | 49 | 6.0 | 86 | 80 |
| 7c | 4-OCH3 | 420 | 58 | 5.0 | 89 | 84 |
| 7d | 3-OH | 540 | 37 | 7.0 | 82 | 77 |
| 7e | 2,4-OH | 420 | 41 | 5.5 | 85 | 76 |
| 7f | 4-F | 420 | 48 | 5.0 | 84 | 84 |
| 7g | 2-Br | 480 | 39 | 6.0 | 82 | 80 |
| 7h | 2-Cl | 520 | 34 | 7.0 | 80 | 84 |
| 7i | 2,4-F | 480 | 39 | 5.0 | 86 | 96 |
| 7j | 4-Cl | 520 | 31 | 7.0 | 84 | 74 |
| 7k | 4-NO2 | 520 | 35 | 7.5 | 85 | 69 |
Assignments of selected characteristic IR bands of the steroidal bis thiocarbazone derivatives (7a–k) provided useful information about the structure of the compounds. All the compounds exhibited a characteristic strong absorption in the region 3229–3309 cm−1 due to the v (N–H) stretching vibration. Intense absorption bands in the regions 1153–1171 cm−1 and 1503–1533 cm−1 were attributed to the v (CS) and v (CN) stretching vibrations, which also confirms the formation of the desired thiocarbazones in all the compounds. All compounds showed a sharp peak at 1731–1735 cm−1, attributable to the ester carbonyl and another sharp peak at 1707–1709 cm−1, due to the steroidal ketones carbonyl (C-7), which also confirms the formation of the monosubstituted compounds. Further evidence of the structure of steroidal bis thiocarbazones was obtained from the 1H NMR spectra. The signal between δ 8.59 and δ 9.27 ppm can be assigned to the protons of the NHN. The signals due to the other NH protons were observed at δ 8.56–9.45 ppm. In addition, the singlet peaks at 0.64, 1.17–1.21 ppm and the doublet at 0.82–0.89 ppm were characteristics of the methyl group of the steroidal structure. The singlet at 3.58 ppm was assigned to the protons of COOCH3. Mass spectra showed the expected molecular peaks at high intensity.
The preliminary results of antibacterial assays indicated that some of the compounds exhibited a moderate to good activity against Gram-negative bacterium (P. seruginosa) and Gram-positive bacteria (S. aureus and B. subtilis). As shown in Table 3, compounds 7b, 7i–k exert no potent inhibition activity against any of the bacterial strains tested. Notably, compounds 7d, 7e and 7h were effective against the Gram-positive S. aureus and compounds 7a, 7c, 7f and 7g were significantly more active against S. aureus than the standard reference drug, amoxicillin. The minimal inhibitory concentration (MIC) values were 0.5–2 μg/mL (7a, 7c, 7f and 7g) and 16 μg/mL (7d, 7e and 7h). The activities of compounds 7a, 7c, 7d, 7f and 7g were found similar to that of amoxicillin against B. subtilis. Most of the compounds showed poor activity against the Gram-negative E. coli and P. aeruginosa. A rapid glance at Table 4 shows that the methoxy derivative 7c was effective at inhibiting Gram-positive S. aureus and B. subtilis. These results reveal that the electron-donating substituent group in these compounds plays a vital role in the inhibitory activity towards Gram-positive bacteria strains. Monosubstituted halogen compounds (except 7j) showed greater antibacterial activity, whereas disubstituted halogen compounds showed weaker antibacterial activity. With respect to P. aeruginosa, only compound 7f showed strong inhibition. On the other hand, only 7f–h showed moderate inhibitions for E. coli.
Antibacterial activity of compounds 7a–k (unit, mm).
| Compounds | Zone of inhibition (mm)a,b | |||
| E. coli | P. aeruginosa | B. subtilis | S. aureus | |
| 7a | – | – | 20.1 ± 0.2 | 25.0 ± 0.1 |
| 7b | – | – | – | – |
| 7c | – | – | 25.5 ± 0.1 | 25.2 ± 0.2 |
| 7d | – | – | 20.5 ± 0.1 | 20.5 ± 0.3 |
| 7e | – | – | 16.1 ± 0.3 | 20.5 ± 0.2 |
| 7f | 15.2 ± 0.2 | 21.1 ± 0.3 | 20.1 ± 0.2 | 25.0 ± 0.3 |
| 7g | 11.9 ± 0.3 | – | 20.4 ± 0.3 | 25.0 ± 0.4 |
| 7h | 11.9 ± 0.4 | – | – | 20.2 ± 0.5 |
| 7i | – | – | – | – |
| 7j | – | – | – | – |
| 7k | – | – | – | – |
| Amoxicillin | 22.9 ± 0.4 | 23.5 ± 0.2 | 25.9 ± 0.4 | 23.5 ± 0.3 |
| Ciprofloxacin | 25.3 ± 0.5 | 31.4 ± 0.4 | 30.0 ± 0.3 | 29.6 ± 0.5 |
| DMSO | – | – | – | – |
a Zones of inhibition less than 10 mm are not shown in the table.
b Reference and compounds (40 μg/mL).
Antibacterial activity of compounds 7a–k.
| Compounds | B. subtilis | S. aureus | E. coli | P. aeruginosa | ||||
| MIC | IC50 | MIC | IC50 | MIC | IC50 | MIC | IC50 | |
| 7a | 16 | 9 | 1 | 0.88 | > 128 | > 64 | > 128 | > 64 |
| 7b | > 128 | > 64 | > 128 | > 64 | > 128 | > 64 | > 128 | > 64 |
| 7c | 1 | 0.86 | 2 | 1.1 | > 128 | > 64 | > 128 | > 64 |
| 7d | 16 | 8.1 | 16 | 8.2 | > 128 | > 64 | > 128 | > 64 |
| 7e | 64 | 35.8 | 16 | 8.5 | > 128 | > 64 | > 128 | > 64 |
| 7f | 8 | 4.7 | 1 | 0.69 | 32 | 19.17 | 16 | 8.65 |
| 7g | 16 | 8.2 | 0.5 | 0.28 | 128 | 70.5 | 128 | > 64 |
| 7h | > 128 | > 64 | 16 | 8.3 | 64 | 48.5 | > 128 | > 64 |
| 7i | > 128 | > 64 | > 128 | > 64 | > 128 | > 64 | > 128 | > 64 |
| 7j | > 128 | > 64 | > 128 | > 64 | > 128 | > 64 | > 128 | > 64 |
| 7k | > 128 | > 64 | > 128 | > 64 | > 128 | > 64 | > 128 | > 64 |
| Amoxicillin | 2 | 1 | 16 | 6.4 | 16 | 7.3 | 4 | 2.5 |
| Ciprofloxacin | 0.128 | 0.1 | 3.12 | 1.82 | 0.25 | 0.18 | 0.5 | 0.41 |
Further, the determination of MIC and 50% inhibition of bacteria growth (IC50) of the active compounds suggested that the fluorine-containing derivative 7f showed the maximum activity against most of the strains tested, with MIC of 32 μg/mL for E. coli, 8 μg/mL for B. subtilis, 16 μg/mL for P. aeruginosa, and 1 μg/mL for S. aureus. Compounds 7a, 7c, 7f and 7g demonstrated notable activity, with MIC values ranging from 0.5 to 2 μg/mL and IC50 from 0.28 to 1.1 μg/mL against S. aureus, showing their potential to become a drug against the drug-resistant S. aureus. Compound 7c was as potent as the standard reference drug amoxicillin against B. subtilis. The importance of this work lies in the possibility that the new compounds might be more effective against bacteria. A thorough investigation regarding the structure–activity relationship, toxicity and the biological effects may allow the design of more potent antibacterial agents for therapeutic use.
3 Conclusion
In conclusion, a microwave-assisted solvent-free synthetic procedure in the presence of neutral aluminum oxide was developed for the preparation of asymmetric chenodeoxycholic acid bis thiocarbazones. The present method has many advantages compared to the conventional method, such as shorter reaction times, high yields and compliance with green chemistry protocols. Compound 7c exhibited promising antibacterial activity similar to that of the standard drug amoxicillin against B. subtilis, indicating that this compound is a promising antibacterial compound for further research. Compounds 7d, 7e and 7h exhibited potency similar to that of amoxicillin against Gram-positive S. aureus, and compounds 7a, 7c, 7f and 7g were found to be very promising and almost comparable to amoxicillin, indicating that they can serve as important pharmacophores for the design of new leading antibacterial agents. Studies to establish their in vitro efficacy and safety are being planned for their further development.
4 Experimental
The melting points were measured with a micro-melting point apparatus and are uncorrected. IR spectra were recorded on 1700 PerkinElmer FTIR using KBr disks. 1H NMR spectra were recorded on a Varian INOVA 400 MHz spectrometer using CDCl3 or DMSO-d6 as a solvent and TMS as an internal standard. Mass spectra were determined with a Finnigan LCQ-DECA instrument. Elemental analysis (C, H, and N) was performed with a Carlo Erba EA1106 auto analyzer and the results were within ± 0.4% of the theoretical values. All the experiments were monitored by thin layer chromatography (TLC) on pre-coated silica gel plates. Optical rotation was measured on a Wzz-2B polarimeter. All the reactions were performed in a commercial microwave apparatus (XH-100A, 100–1000 W, Beijing Xianghu Science and Technology Development Co. Ltd, Beijing, China). All the solvents were purified before use. Pyridinium chlorochromate (PCC) and methyl (5β)-3,7-dihydroxycholan-24-oate 5 were prepared according to the methods reported [31,32].
4.1 Preparation of substituted benzaldehyde thiocarbohydrazones (3a–k)
The aromatic aldehyde (10 mmol) in ethanol (40 mL) was added dropwise to a solution of thiocarbohydrazide (15 mmol) in H2O (50 mL) and acetic acid (1 mL). The mixture was refluxed in the microwave oven at 300 W for 15 min. The reaction was monitored by TLC until completion. The precipitate was collected by filtration and washed with water (3 × 10 mL). The resulting solid was recrystallised from ethanol to give the product (Scheme 2). The analytical data for 3i are given below. The melting points of benzaldehyde thiocarbohydrazones 3a–k are shown in Table 5.
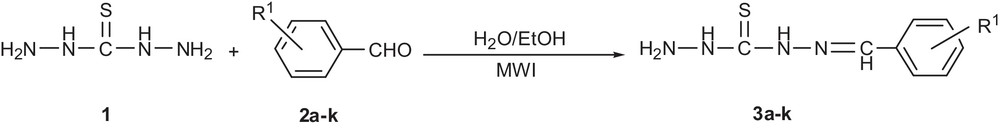
Synthetic protocol of compounds 3a–k.
The melting points of substituted benzaldehyde thiocarbohydrazones 3a–k.
| Product | Formula | R1 | Mp (°C) | Lit. Mp (°C) |
| 3a | C8H10N4S | H | 197–198 | 198 [33] |
| 3b | C8H10N4OS | 2-OH | 218–219 | 218 [33] |
| 3c | C9H12N4OS | 4-OCH3 | 198–199 | 198–200 [34] |
| 3d | C8H10N4OS | 3-OH | 201–202 | 200–201[28] |
| 3e | C8H10N4O2S | 2,4-OH | 214–215 | 215–216 [35] |
| 3f | C8H9FN4S | 4-F | 208–209 | 208–209 [28] |
| 3g | C8H9BrN4S | 2-Br | 200–201 | 200–201 [35] |
| 3h | C8H9ClN4S | 2-Cl | 209–210 | 210 [33] |
| 3i | C8H8F2N4S | 2,4-F | 169–170 | – |
| 3j | C8H9ClN4S | 4-Cl | 211–212 | 211–212 [28] |
| 3k | C8H9N5O2S | 4-NO2 | 204–206 | 204–206 [36] |
4.1.1 Compound 3i
White crystal. Yield 79%. Mp 169–170 °C (MeOH–H2O). IR (KBr, cm−1): 3409, 3356, 3265, 2930, 2791, 1615, 1564, 1507, 1429, 1283, 1253, 1141, 1077, 1018, 848, 611. 1H NMR (DMSO-d6, 400 MHz): δ 11.53 (s, 1H, NH), 9.96 (s, 1H, NH), 8.45–8.39 (m, 1H, ArH), 8.17 (s, 1H, CH), 7.33–7.27 (m, 1H, ArH), 7.15 (dd, J = 6.4 and 8.4 Hz, 1H, ArH), 4.88 (s, 2H, NH2). ESI–MS m/z (%): 231 ([M + 1]+, 100). Anal. calcd for C8H8F2N4S: C, 41.73; H, 3.50; N, 24.33. Found: C, 41.78; H, 3.48; N, 24.35%.
4.2 Synthesis of methyl (5β)-3,7-dioxocholan-24-oate (6) [37]
Pyridinium chlorochromate (PCC) (3.03 mmol) was added to a solution of methyl (5β)-3,7-dihydroxycholan-24-oate 5 (0.25 g, 0.615 mmol) in dried CH2Cl2 (20 mL) at room temperature. The reaction was completed in 12 h. The solvent was removed under reduced pressure, and the crude product was purified by chromatography on silica gel using ethyl acetate to give a white crystal. Yield 86%. Mp 155–156 °C (ethyl acetate) (lit. [38] Mp 154–156 °C), [α]D = −21.5 (c 0.12, CH2Cl2). IR (KBr, cm−1): 2953, 2873, 1709, 1403, 1375, 1214, 1174, 1010, 966. 1H NMR (CDCl3, 400 MHz): δ 3.67 (s, 3H, COOCH3), 1.31 (s, 3H, 19–CH3), 0.93 (d, J = 6.4 Hz, 3H, 21–CH3), 0.69 (s, 3H, 18–CH3). ESI–MS m/z (%): 827 ([2M + 23]+, 100). Anal. calcd for C25H38O4: C, 74.48; H, 9.54. Found. C, 74.51; H, 9.53%.
4.3 Microwave procedure for the preparation of bis thiocarbazones (7a–k)
The steroidal diketone methyl (5β)-3,7-dioxocholan-24-oate (6) (0.1 mmol) and the substituted benzaldehyde thiocarbohydrazones 3a–k (0.1 mmol) and natural aluminum oxide (0.3 g) were placed in a porcelain mortar, and concentrated HCl (two drops) was added. After grinding, the mixture was put in a round-bottom flask (25 mL) and irradiated in a microwave oven for 5.0–7.5 min at 350 W. The reaction mixture was cooled to room temperature, dissolved in DMSO and filtered. The product was recrystallised from ethanol in 80–89% yields. The physical and spectral data of the compounds 7a–k are as follows.
4.3.1 Methyl (5β)-3-[2-[[(phenylideneamin)amino]thioxomethyl]hydrazinylidene]-7-oxocholan-24-oate (7a)
White crystal. Yield 84%. Mp 125–126 °C (EtOH), [α]D = −108.7 (c 0.10, CH2Cl2). IR (KBr, cm−1): 3263, 2945, 2871, 1735, 1708, 1630, 1517, 1380, 1235, 1215, 1169, 1099, 1021, 757. 1H NMR (DMSO-d6, 400 MHz): δ 11.72 (s, 1H, NH), 10.60 (s, 1H, NH), 8.10 (s, 1H, CH), 7.72 (s, 2H, ArH), 7.43 (t, J = 5.6 Hz, 3H, ArH), 3.58 (s, 3H, COOCH3), 1.21 (s, 3H, 19–CH3), 0.89 (d, J = 6.4 Hz, 3H, 21–CH3), 0.64 (s, 3H, 18–CH3). ESI–MS m/z (%): 1157 ([2M + 1]+, 100). Anal. calcd for C33H46N4O3S: C, 68.48; H, 8.01; N, 9.68. Found. C, 68.45; H, 8.04; N, 9.69%.
4.3.2 Methyl (5β)-3-[2-[[(2-hydroxyphenylideneamin)amino]thioxomethyl]hydrazinylidene]-7-oxocholan-24-oate (7b)
White crystal. Yield 86%. Mp 173–174 °C (EtOH), [α]D = −58.9 (c 0.10, CH2Cl2). IR (KBr, cm−1): 3265, 3128, 2949, 1735, 1707, 1615, 1533, 1488, 1438, 1379, 1272, 1226, 1199, 1153, 1038, 958. 1H NMR (DMSO-d6, 400 MHz): δ 11.74 (s, 1H, NH), 11.64 (s, 1H, OH), 10.89 (d, J = 9.2 Hz, 1H, NH), 8.69 (s, 1H, CH), 7.35 (s, 1H, ArH), 7.29 (t, J = 8.0 Hz, 2H, ArH), 6.91 (t, J = 4.4 Hz, 1H, ArH), 3.58 (s, 3H, COOCH3), 1.21 (s, 3H, 19–CH3), 0.89 (d, J = 6.4 Hz, 3H, 21–CH3), 0.64 (s, 3H, 18–CH3). ESI–MS m/z (%): 1189 ([2M + 1]+, 100). Anal. calcd for C33H46N4O4S: calcd. C, 66.64; H, 7.79; N, 9.42. Found. C, 66.59; H, 7.81; N, 9.43%.
4.3.3 Methyl (5β)-3-[2-[[(4-methoxyphenylideneamin)amino]thioxomethyl]hydrazinylidene]-7-oxocholan-24-oate (7c)
White crystal. Yield 89%. Mp 120–121 °C (EtOH), [α]D = −154.8 (c 0.10, CH2Cl2). IR (KBr, cm−1): 3253, 2944, 1735, 1708, 1605, 1505, 1379, 1301, 1250, 1169, 1103, 1027, 931. 1H NMR (DMSO-d6, 400 MHz): δ 11.63 (s, 1H, NH), 10.51 (s, 1H, NH), 8.05 (s, 1H, CH), 7.65 (t, J = 4.4 Hz, 2H, ArH), 7.01 (d, J = 8.4, 2H, ArH), 3.80 (s, 3H, Ar–OCH3), 3.58 (s, 3H, COOCH3), 1.21 (s, 3H, 19–CH3), 0.89 (d, J = 6.4 Hz, 3H, 21–CH3), 0.64 (s, 3H, 18–CH3). ESI–MS m/z (%): 631 ([M + 23]+, 100). Anal. calcd for C34H48N4O4S: C, 67.07; H, 7.95; N, 9.20. Found. C, 67.00; H, 6.97; N, 9.26%.
4.3.4 Methyl (5β)-3-[2-[[(3-hydroxyphenylideneamin)amino]thioxomethyl]hydrazinylidene]-7-oxocholan-24-oate (7d)
White crystal. Yield 82%. Mp 144–145 °C (EtOH), [α]D = −100.7 (c 0.10, CH2Cl2). IR (KBr, cm−1): 3272, 2946, 2870, 1707, 1581, 1522, 1451, 1380, 1225, 1170, 955, 784. 1H NMR (DMSO-d6, 400 MHz): δ 11.36 (s, 1H, NH), 10.57 (s, 1H, NH), 9.62 (d, J = 5.2 Hz, 1H, OH), 8.02 (s, 1H, CH), 7.28–7.18 (m, 1H, ArH), 7.14–7.07 (m, 2H, ArH), 6.82 (d, J = 7.6 Hz, 1H, ArH), 3.58 (s, 3H, COOCH3), 1.21 (s, 3H, 19–CH3), 0.89 (d, J = 6.4 Hz, 3H, 21–CH3), 0.64 (s, 3H, 18–CH3). ESI–MS m/z (%): 1189 ([2M + 1]+, 100). Anal. calcd for C33H46N4O4S: C, 66.64; H, 7.79; N, 9.42. Found. C, 66.68; H, 7.75; N, 9.45%.
4.3.5 Methyl (5β)-3-[2-[[(2,4-dihydroxyphenylideneamin)amino]thioxomethyl]hydrazinylidene]-7 -oxocholan-24-oate (7e)
White crystal. Yield 85%. Mp 222–223 °C (EtOH), [α]D = −97.4 (c 0.10, CH2Cl2). IR (KBr, cm−1): 3309, 3233, 3149, 2950, 1732, 1709, 1624, 1515, 1378, 1330, 1208, 1171, 1111, 979, 885. 1H NMR (DMSO-d6, 400 MHz): δ 11.76 (s, 1H, NH), 11.55 (s, 1H, OH), 10.75 (s, 1H, NH), 9.92 (s, 1H, OH), 8.56 (s, 1H, CH), 7.12 (s, 1H, ArH), 6.34 (d, J = 7.2 Hz, 1H, ArH), 6.28 (d, J = 2.0 Hz, 1H, ArH), 3.58 (s, 3H, COOCH3), 1.21 (s, 3H, 19–CH3), 0.89 (d, J = 6.4 Hz, 3H, 21–CH3), 0.64 (s, 3H, 18–CH3). ESI–MS m/z (%): 611 ([M + 1]+, 100). Anal. calcd for C33H46N4O5S: C, 64.89; H, 7.59; N, 9.17. Found. C, 64.92; H, 7.57; N, 9.15%.
4.3.6 Methyl (5β)-3-[2-[[(4-fluorophenylideneamin)amino]thioxomethyl]hydrazinylidene]-7-oxocholan-24-oate (7f)
White crystal. Yield 84%. Mp 124–125 °C (EtOH), [α]D = −97.9 (c 0.10, CH2Cl2). IR (KBr, cm−1): 3229, 2945, 2870, 1735, 1708, 1601, 1503, 1380, 1232, 1156, 1097, 1017, 896, 835. 1H NMR (DMSO-d6, 400 MHz): δ 11.72 (s, 1H, NH), 10.61 (s, 1H, NH), 8.08 (s, 1H, CH), 7.77 (t, J = 12 and 5.2 Hz, 2H, ArH), 7.29 (t, J = 8.8 and 8.8 Hz, 2H, ArH), 3.58 (s, 3H, COOCH3), 1.21 (s, 3H, 19–CH3), 0.89 (d, J = 6.4 Hz, 3H, 21–CH3), 0.64 (s, 3H, 18–CH3). ESI–MS m/z (%): 1193 ([2M + 1]+, 100). Anal. calcd for C33H45FN4O3S: C, 66.41; H, 7.60; N, 9.39. Found. C, 66.48; H, 7.58; N, 9.37%.
4.3.7 Methyl (5β)-3-[2-[[(2-bromophenylideneamin)amino]thioxomethyl]hydrazinylidene]-7-oxocholan-24-oate (7g)
White crystal. Yield 82%. Mp 129–130 °C (EtOH), [α]D = −79.4 (c 0.10, CH2Cl2). IR (KBr, cm−1): 3236, 2944, 2869, 1734, 1708, 1589, 1516, 1466, 1435, 1378, 1228, 1203, 1168, 1100, 1021, 821, 757. 1H NMR (DMSO-d6, 400 MHz): δ 11.80 (s, 1H, NH), 10.68 (s, 1H, NH), 8.47 (s, 1H, CH), 8.12 (s, 1H, ArH), 7.67 (d, J = 8.0 Hz, 1H, ArH), 7.47–7.41 (m, 1H, ArH), 7.37–7.33 (m, 1H, ArH), 3.58 (s, 3H, COOCH3), 1.21 (s, 3H, 19–CH3), 0.89 (d, J = 6.4 Hz, 3H, 21–CH3), 0.64 (s, 3H, 18–CH3). ESI–MS m/z (%): 657 ([M + 1]+, 100), 658 ([M + 2]+, 93). Anal. calcd for C33H45BrN4O3S: C, 60.26; H, 6.90; N, 8.52. Found. C, 60.30; H, 6.87; N, 8.50%.
4.3.8 Methyl (5β)-3-[2-[[(2-chlorophenylideneamin)amino]thioxomethyl]hydrazinylidene]-7-oxocholan-24-oate (7h)
White crystal. Yield 80%. Mp 127–128 °C (EtOH), [α]D = −86.7 (c 0.10, CH2Cl2). IR (KBr, cm−1): 3239, 2945, 2870, 1735, 1708, 1594, 1516, 1468, 1437, 1379, 1228, 1204, 1169, 1049, 893. 1H NMR (DMSO-d6, 400 MHz): δ 11.79 (s, 1H, NH), 10.68 (s, 1H, NH), 8.51 (s, 1H, CH), 8.19 (s, 1H, ArH), 7.58–7.48 (m, 1H, ArH), 7.45–7.42 (m, 2H, ArH), 3.58 (s, 3H, COOCH3), 1.21 (s, 3H, 19–CH3), 0.89 (d, J = 6.4 Hz, 3H, 21–CH3), 0.64 (s, 3H, 18–CH3). ESI–MS m/z (%): 1225 ([2M + 1]+, 100), 1226 ([2M + 2]+, 42). Anal. calcd for C33H45ClN4O3S: C, 64.63; H, 7.40; N, 9.14. Found. C, 64.60; H, 7.41; N, 9.17%.
4.3.9 Methyl (5β)-3-[2-[[(2,4-difluorophenylideneamin)amino]thioxomethyl]hydrazinylidene]-7-oxocholan-24-oate (7i)
White crystal. Yield 86%. Mp 126–127 °C (EtOH), [α]D = −102.7 (c 0.10, CH2Cl2). IR (KBr, cm−1): 3253, 2948, 2872, 1734, 1708, 1614, 1523, 1499, 1431, 1381, 1279, 1230, 1171, 1140, 1100, 965, 850. 1H NMR (DMSO-d6, 400 MHz): δ 11.80 (s, 1H, NH), 10.68 (s, 1H, NH), 8.72 (s, 1H, CH), 8.19 (d, J = 10.4 Hz, 1H, ArH), 7.41–7.33 (m, 1H, ArH), 7.22 (d, J = 8.0 Hz, 1H, ArH), 3.58 (s, 3H, COOCH3), 1.21 (s, 3H, 19–CH3), 0.82 (d, J = 6.0 Hz, 3H, 21–CH3), 0.64 (s, 3H, 18–CH3). ESI–MS m/z (%): 615 ([M + 1]+, 100); Anal. calcd for C33H44F2N4O3S: C, 64.47; H, 7.21; N, 9.11. Found. C, 64.50; H, 7.20; N, 9.10%.
4.3.10 Methyl (5β)-3-[2-[[(4-chlorophenylideneamin)amino]thioxomethyl]hydrazinylidene]-7-oxocholan-24-oate (7j)
White crystal. Yield 84%. Mp 131–132 °C (EtOH), [α]D = −84.2 (c 0.10, CH2Cl2). IR (KBr, cm−1): 3250, 2948, 2864, 1731, 1708, 1633, 1591, 1521, 1489, 1241, 1209, 1168, 1086, 1016, 828. 1H NMR (DMSO-d6, 400 MHz): δ 11.75 (s, 1H, NH), 10.49 (s, 1H, NH), 8.49 (s, 1H, CH), 7.71 (dd, J = 10.4 and 8.8 Hz, 2H, ArH), 7.56–7.44 (m, 2H, ArH), 3.58 (s, 3H, COOCH3), 1.17 (s, 3H, 19–CH3), 0.82 (d, J = 6.0 Hz, 3H, 21–CH3), 0.64 (s, 3H, 18–CH3). ESI–MS m/z (%): 613 ([M + 1]+, 100), 614 ([M + 2]+, 43). Anal. calcd for C33H45ClN4O3S: C, 64.63; H, 7.40; N, 9.14. Found. C, 64.68; H, 7.42; N, 9.12%.
4.3.11 Methyl (5β)-3-[2-[[(4-nitrophenylideneamin)amino]thioxomethyl]hydrazinylidene]-7-oxocholan-24-oate (7k)
White crystal. Yield 85%. Mp 129–130 °C (EtOH), [α]D = −135.7 (c 0.11, CH2Cl2). IR (KBr, cm−1): 3238, 2947, 2871, 1735, 1708, 1589, 1519, 1484, 1439, 1380, 1241, 1207, 1171, 1009, 822. 1H NMR (DMSO-d6, 400 MHz): δ 11.76 (s, 1H, NH), 10.65 (s, 1H, NH), 8.48 (s, 1H, CH), 7.83–7.61 (m, 4H, ArH), 3.58 (s, 3H, COOCH3), 1.17 (s, 3H, 19–CH3), 0.82 (d, J = 6.4 Hz, 3H, 21–CH3), 0.64 (s, 3H, 18–CH3). ESI–MS m/z (%): 1247 ([2M + 1]+, 100). Anal. calcd for C33H45N5O5S: C, 64.54; H, 7.27; N, 11.23. Found. C, 64.59; H, 7.25; N, 7.29%.
4.4 Conventional procedure for the preparation of bis thiocarbazones (7a–k)
The steroidal diketone methyl (5β)-3,7-dioxocholan-24-oate (6) (0.1 mmol) and the substituted benzaldehyde thiocarbohydrazones 3a–k (0.1 mmol) were dissolved in ethanol (15 mL). After complete dissolution, two drops of HCl were added. The mixture was stirred for 420–540 min at 80 °C. After cooling, the products were filtered and recrystallized form ethanol in 31–58% yields.
4.5 Bioactivity
The in vitro antimicrobial activity of the structurally promising steroidal bis thiocarbazones 7a–k against two strains of Gram-positive bacteria (B. subtilis and S. aureus) and two strains of Gram-negative (E. coli and P. aeruginosa) were investigated using disc-diffusion method and microdilution method in comparison to the reference drugs amoxicillin and ciprofloxacin. Dimethylformamide was used as the negative control.
4.5.1 Disc-diffusion assay
The compounds were tested for their antibacterial activities by disc-diffusion method using a nutrient agar medium [39,40]. In the disc-diffusion method, serial dilutions of the test compounds were prepared to final concentrations of 40, 20, 10, 5 and 0 μg/mL in DMSO. Sterile paper discs (6 mm) were then placed onto the nutrient agar medium and 20 μL (40, 20, 10, 5 and 0 mg/mL) of the sample were added per disc in five replications. Then, the paper discs impregnated with the solutions of the tested compounds in DMSO were placed on the surface of the media inoculated with the microorganism. The plates were incubated at 37 °C for 16–18 h (Table 3). The IC50 was computed from the diameter values of growth inhibition zones.
4.5.2 Microdilution assay
The MIC were determined by microdilution, both methods [41] using standard inoculums of 2 × 106 CFU/mL. Serial dilutions of the test compounds were prepared to final concentrations of 256, 128, 64, 32, 16, 8, 4, 2, 1, 0.5, 0.25 and 0 μg/mL in DMSO. To each tube was added 1 mL of a bacterial fluid (0.5 McFarland standard). The MIC that inhibits the visible growth after 18 h was determined visually after incubation for 18 h at 37 °C (Table 4).
Acknowledgments
This research was financially supported by the Science and Technology Department of Sichuan Province (No. 2011JY0035), the Fundamental Research Funds of the Central University, Southwest University for Nationalities (No. 13NZYQN12) and the Postgraduate Degree Construction Project of Southwest University for Nationalities of P.R. China (No. 2013XWD-S0703).