

1 Introduction
Calix[4]resorcinarenes (resorcinarenes) are widespread cyclic tetrameric host compounds for ions, sugars and organic molecules; as a consequence they are used as starting materials for a variety of cavitands or other macrocyclic host molecules[1]. They have also been applied as stationary phases in HPLC [2] and chiral capillary gas chromatography for the separation of positional isomers of substituted benzenes [3]. Furthermore, they can exhibit a liquid-crystalline behavior through the appropriate choice of the R groups on the resorcinarene [4]. Fig. 1 represents the general procedure for the formation of calix[4]resorcinarenes by cyclocondensation of resorcinol and aldehydes.
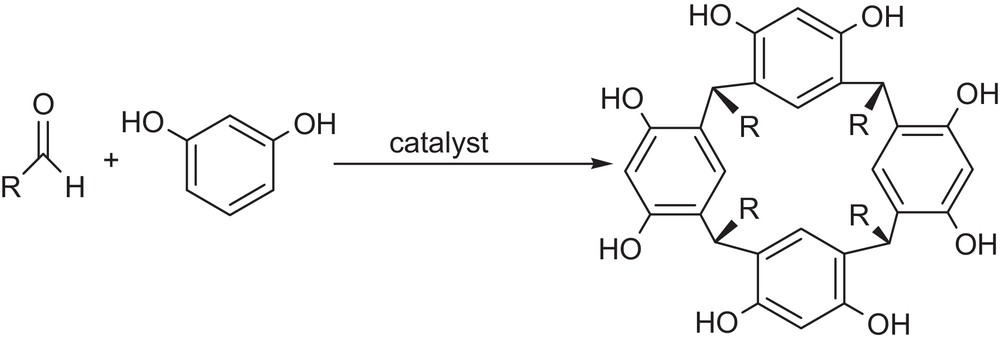
Synthesis of calix[4]resorcinarenes by cyclocondensation of resorcinol and aldehydes.
The most common synthetic method still involves mineral acid catalysis of this condensation in hot alcohol solution [5], in which the use of large quantities of concentrated HCl leads to excessive waste streams that are environmentally unfriendly and expensive to deal with. A solvent-free synthesis method of calix[4]resorcinarenes using p-TsOH as the catalyst has also been reported [6]. This convincing green approach was unsuccessful because the longer the alkyl chain of the aldehyde, the lower the reaction yield. Classical Lewis acids like BF3.OEt2, AlCl3 and SnCl4 have been used only for the synthesis of aromatic aldehyde-derived resorcinarenes [7]. Unfortunately, this method has several disadvantages such as the use of large quantities of these Lewis acids to obtain good yields of the product and the sensitivity of these catalysts to water, which results in acidic and metal oxide by-products upon aqueous work-up. More recently, triflate-based catalysts such as ytterbium(III) triflate [8] and bismuth(III) triflate [9] have been introduced for this condensation. Expensive preparation and corrosive starting material to prepare triflate salt could be one of the disadvantages of this method. The use of lanthanide sulfonate [10] and the microwave-assisted synthesis of resorcinarene in the presence of HCl and tungstophosphoric acid (TPA) has also been reported [11]. Therefore, the development of new synthetic methods would be of special interest.
Molecular iodine as a mild Lewis acid has received considerable attention as an inexpensive, non-toxic, readily available and environmentally friendly catalyst in organic and pharmaceutical syntheses [12]. It is also highly tolerant to air as well as to moisture. Recent studies indicate that iodine can be effectively used as a catalyst for acetylation [13], protections and deprotections, oxidations, three-component synthesis of protected homoallylic amines [14], direct oxidative conversion of alkyl halides into nitriles, synthesis of bis(indolyl) methanes [15], alkylation of active methylene compounds [16], and synthesis of amidophenol [17]. We now report the utility of iodine as an efficient catalyst for the condensation of aldehydes and resorcinol to give the corresponding resorcinarenes in excellent yields.
2 Results and discussion
Initially the reaction between pentanal and resorcinol was used as a model reaction. To optimize the catalyst's loading, amounts of 0, 5, 10, and 20 mol% of iodine were tested, respectively. The results are summarized in Table 1. A 20 mol% loading of iodine was sufficient to obtain the best yield of resorcinarene 1a (Table 1, entry 4). To optimize the reaction temperature, the synthesis of compound 1a was carried out in ethanol at room temperature, 30, 50, and 78 °C, respectively (Table 2). We found that below the reflux temperature of ethanol, no reaction occurred. Therefore, the desired reaction temperature should be 78 °C (Table 2, entry 4).
The effect of different amounts of iodine for the synthesis of 1a (R = C4H9).
| Isolated yield [%] | Iodine [mmol%] | Entry |
| — | 0 | 1 |
| 73 | 5 | 2 |
| 80 | 10 | 3 |
| 95 | 20 | 4 |
Optimization of temperature for the synthesis of 1a (R = C4H9).
| Isolated yield [%] | Temp. [°C] | Entry |
| — | r.t. | 1 |
| — | 30 | 2 |
| — | 50 | 3 |
| 95 | 78 | 4 |
We carefully analyzed the 1H NMR spectra of 1, 2, 3 (aliphatic) and 4, 5, 6 (aromatic). The condensation reaction of resorcinol with aromatic aldehydes in the presence of iodine formed a mixture of diastereomers (Fig. 2). With benzaldehyde, the product was obtained as a mixture of diastereomers, all-cis (rccc, C4v) isomer (a) and the cis-trans-trans (rctt, C2v) isomer (b). The 1H NMR spectrum of benzaldehyde-derived resorcinarene showed one singlet at δ = 5.37 for the bridge benzylic protons (4 H) and two singlets at δ = 5.55 and 6.11 for the intra-annular aromatic protons Hin (each 2 H), indicating that the C2v isomer had been produced. An equivalent single resonance for the four ortho protons of resorcinol rings at δ = 6.12 and the four meta protons at δ = 6.33 were consistent with those previously reported for the C4v isomer. Other aromatic aldehydes such as 4-hydroxybenzaldehyde and 4-methoxybenzaldehyde were also tested.
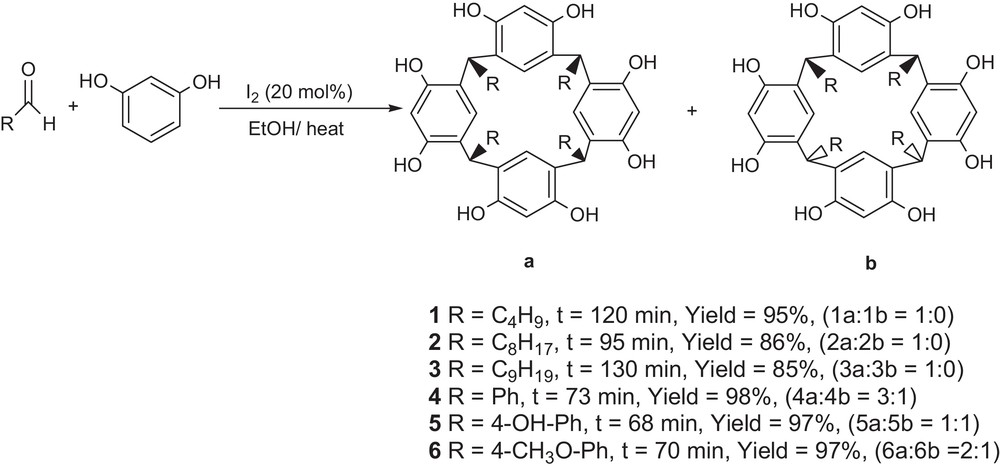
Condensation reaction of resorcinol with aromatic aldehydes in the presence of iodine, affording a mixture of diastereomers.
In both cases, the corresponding resorcinarenes were a mixture of C4v and C2v isomers as well. For the 1H NMR spectrum of resorcinarene 5, one singlet at δ = 5.42 and two singlets at δ = 5.92 and 6.09 indicated the presence of the rctt isomer, while a single resonance for the four ortho protons of resorcinol rings at δ = 6.08 and the four meta protons at δ = 6.31 proved the existence of the rccc configuration. The thermodynamically favored all-cis isomer (via single resonances at δ = 6.11 and 6.30) besides the cis-trans-trans isomer (based on singlet resonances at δ = 5.45, 5.55 and 6.11) were clearly identified in the 1H NMR spectrum of resorcinarene 6. The ratio of diastereomers after completion of the reaction (as judged by the disappearance of the starting materials by TLC) was obtained by integration of the signals of the benzylic hydrogens of the rccc isomer to the signals of the benzylic hydrogens of the rctt isomer. For the resorcinarenes derived from aliphatic substrates, the tetrameric nature of the products was only that of the all-cis (rccc) isomer.
Finally, a comparison was made between the synthesis of resorcinarene 4 and other reported procedures. The results summarized in Table 3 indicate that the present procedure is better than all the earlier reported protocols in terms of availability and cost of the catalyst.
Comparison between various methods and this work for the synthesis of 4aa.
| Entry | Catalyst | Time | Yield(%) | cccc:cttcb | Reference |
| 1 | HCl | 11days | 83 | 32:1 | [5c] |
| 2 | [Yb(H2O)9](OTf)3 | 48 h | 1.4:1 | [8] | |
| 3 | PTSA | 60 min | 1:2c | [6] | |
| 4 | BiCl3 | 75 min | 66 | 1:1.2 | [9] |
| 5 | Yb(TOS)3 | 24 h | 93 | 1:1.2 | [10] |
| 6 | I2 | 75 min | 3:1 | This work |
a Reaction conditions: benzaldehyde (1 mmol), resorcinol (1 mmol), ethanol, catalyst.
b Diastereomeric ratio from 1H NMR.
c Solvent-free conditions.
3 Conclusion
In summary, we have developed an alternative and simple method for the preparation of calix[4]resorcinarenes in the presence of iodine as a catalyst. The advantages of this method include reasonable reaction time, the use of very cheap and easy available catalyst, excellent yields, and simple workup. The reactions could also be carried out in air, since iodine is not moisture sensitive. Consequently, this method has wide scope for further applications.
4 Experimental
4.1 General remarks
All the chemicals were purchased from Merck Chemicals and used without further purification. The melting points were taken in open capillary tubes with an Electrothermal 9100 Apparatus. FT–IR (KBr) spectra were recorded on an ABB FT-IR FTLA 2000 spectrometer. 1H NMR spectra were run on a Bruker DRX-300 (300 MHz) AVANCE instrument using TMS as an internal standard and DMSO-d6 as a solvent. The chemical shifts (δ) are reported in ppm relative to TMS as an internal standard and J values are given in Hz. 13C spectra were recorded at 75 MHz. Mass spectra were recorded using a Hewlett Packard 5973 mass spectrometer.
4.2 Typical procedure for the synthesis of resorcinarenes
Iodine (20 mol%) was added to a solution of resorcinol (1 mmol) and of an aldehyde (1 mmol) in 2 mL of absolute ethanol. The solution was stirred at 78 °C. After a given period of time, the solution was cooled and poured into 10 mL of an ice-cold saturated sodium thiosulfate aqueous solution. The resulting precipitate was collected by filtration and washed with water. The precipate was dried at 90 °C and identified.
4.2.1 Spectroscopic data of 2,4,8,20-tetrabutylpentacyclo[19.3.1.1.1.1]octacosa 1(25),3,5,7(28),9,11,13(27),15,17,19(26),21,23-dodecaene-4,6,10,12,16,18,22,24-octol (1a)
1H NMR (DMSO-d6) δ (ppm): 0.79 (t, 12H, J = 7.33 Hz, CH3), 1.08 (m, 8H, CH2CH3), 1.28 (quin, 8H, J = 6.84 Hz, CH2CH2CH3), 2.01 (q, 8H, J = 6.73, ArCHCH2), 4.19 (t, 4H, J = 7.69 Hz, methine) 6.13 (s, 4H, ArH, ortho to OH), 7.14 (s, 4H, ArH, meta to OH), 8.85 (s, 8H, 8OH). 13C NMR (DMSO-d6): δ (ppm) = 14.1, 22.2, 30.1, 32.9, 33.6, 75.5, 102.3, 123.1, 125.0, 151.6. EI–MS, m/z = 712 (M+).
4.2.2 Spectroscopic data of 2,4,8,20-tetraoctylpentacyclo[19.3.1.1.1.1]octacosa 1(25),3,5,7(28),9,11,13(27),15,17,19(26),21,23-dodecaene-4,6,10,12,16,18,22,24-octol (2a)
1H NMR (DMSO-d6) δ (ppm): 0.81 (t, 12H, J = 7.33 Hz, CH3), 1.20 (br, 48H, (CH2)6CH3), 1.96 (br, 8H, J = 6.84 Hz, CHCH2), 4.23 (t, 4H, J = 7.69 Hz, methine) 6.14 (s, 4H, ArH, ortho to OH), 7.08 (s, 4H, ArH, meta to OH), 8.85 (s, 8H, 8OH). 13C NMR (DMSO-d6): δ (ppm) = 14.0, 22.6, 29.2.2, 22.4, 27.6, 31.6, 31.9, 34.1, 75.5, 102.7, 123.2, 124.8, 151.8. EI–MS, m/z = 938 (M+).
4.2.3 Spectroscopic data of 2,4,8,20-tetranonylpentacyclo[19.3.1.1.1.1]octacosa 1(25),3,5,7(28),9,11,13(27),15,17,19(26),21,23-dodecaene-4,6,10,12,16,18,22,24-octol (3a)
1H NMR (DMSO-d6) δ (ppm): 0.812 (t, 12H, J = 7.33 Hz, CH3), 1.20 (br, 56H, (CH2)7CH3), 1.96 (br, 8H, J = 6.84 Hz, CHCH2), 4.21 (t, 8H, J = 7.69 Hz, methine) 6.15 (s, 4H, ArH, ortho to OH), 7.06 (s, 4H, ArH, meta to OH), 8.87 (s, 8H, 8OH). 13C NMR (DMSO-d6): δ (ppm) =13.9, 14.1, 20.7, 22.1, 27.8, 28.7, 29.1, 29.2, 31.4, 59.8, 102.3, 122.8, 123.7, 151.7. EI–MS, m/z = 993 (M+).
4.2.4 Spectroscopic data of 2,4,8,20-tetraphenylpentacyclo[19.3.1.1.1.1]octacosa 1(25),3,5,7(28),9,11,13(27),15,17,19(26),21,23-dodecaene-4,6,10,12,16,18,22,24-octol (4a/4b)
1H NMR (DMSO-d6) δ (ppm): 5.53(s, 4H, ArCH (C2v)), 5.55 (s, 2H, ArH (C2v) ortho to OH), 5.62(s, 4H, ArCH (C4v)), 6.11 (s, 2H, ArH (C2v) ortho to OH) 6.12 (s, 4H, ArH (C4v), ortho to OH), 6.33 (s, 4H, ArH (C4v), meta to OH), 6.34 (s, 4H, ArH (C2v), meta to OH), 6.58–6.60 (m, 8H, ArH (C2v)), 6.72–6.74 (d, 8H, ArH (C4v)), 6.83 (d, 12 H,, ArH (C2v)), 6.93 (d, 12 H,, ArH (C4v)), 8.44 (s, 4 OH, ArOH (C2v)), 8.55 (s, 4OH (C2v) 8OH (C4v), ArOH)
4.2.5 Spectroscopy data 2,4,8,20-tetra4-hydroxy phenylpentacyclo[19.3.1.1.1.1] octacosa1(25),3,5,7(28),9,11,13(27),15,17,19(26),21,23-dodecaene 4,6,10,12,16,18,22,24-octol (5a/5b)
1H NMR (DMSO-d6) δ (ppm): 5.42 (s, 4H, ArCH (C2v)), 5.52 (s, 4H, ArCH (C4v)), 5.92 (s, 2H, ArH (C2v) ortho to OH), 6.08 (s, 4H, ArH (C4v), ortho to OH), 6.02 (s, 2H, ArH (C2v) ortho to OH), 6.27 (s, 2H, ArH (C2v), meta to OH), 6.31 (s, 4H, ArH (C4v), meta to OH), 6.33 (s, 2H, ArH (C2v) meta to OH), 6.41–6.43 (d, 8H, J = 4.71 Hz, ArH (C4v)), 8.35 (s, 4OH, ArOH (C2v), 8.39 (s, 4OH, ArOH (C2v)), 8.43 (s, 4OH, ArOH (C4v)), 8.66 (s, 4 OH, ArOH (C2v), 8.83 s, 4OH, ArOH (C4v)).
4.2.6 Spectroscopic data of 2,4,8,20-tetra4-methoxy phenylpentacyclo[19.3.1.1.1.1] octacosa1(25),3,5,7(28),9,11,13(27),15,17,19(26),21,23-dodecaene 4,6,10,12,16,18,22,24-octol (6a/6b)
3.63 (s, 12 H CH3 (C2v)), 3.70 (s, 12 H CH3 (C4v)), 5.45 (s, 4H, ArCH (C2v)), 5.55 (s, 2H, ArCH (C2v) ortho to OH), 5.58 (s, 4H, ArCH (C4v)), 6.11 (s, 4H, ArH (C4v), ortho to OH and 2H, ArH (C2v), ortho to OH), 6.28 (s, 2H, ArH (C2v) meta to OH), 6.30 (s, 2H, ArH (C2v), meta to OH and 4H, ArH (C4v)), 6.43–6.45 (d, 8H, J = 4.71 Hz, ArH (C4v)), 6.49–6.51 (d, 8H, J = 4.8 Hz, ArH (C2v)), 6.53–6.55 (d, 8H, J = 4.8 Hz, ArH (C2v)), 6.59–6.61 (d, 8H, J = 4.71 Hz, ArH (C4v)), 8.41 (s, 4 OH, ArOH (C2v), 8.50 (s, 4 OH, ArOH (C2v).