

1 Introduction
The protection and deprotection of the alcohol functional group are of high interest, and have been widely considered because of their basic role in multi-step syntheses. Whereas protons of alcohols have some acidic properties and can be separated in the vicinity of strong bases, hydroxyl functional groups in alcohols can contribute to many unfavorable reactions, even under mild conditions, resulting in side reactions, thus decreasing the reaction yield. Therefore, developing mild and efficient procedures for the protection and deprotection of the alcohols’ proton is an important aim for synthetic chemists. One of the conventional methods used to protect alcohols is their transformation to triphenylmethyl (trityl) ether. The trityl group (Tr) is one of the most advantageous protecting groups, because it can be removed easily. Although the trityl group is stable under neutral or basic conditions, it can be readily removed under mild acidic conditions. The introduction of this group into the hydroxyl functionality has been carried out using usually trityl halides and trityl ethers (TrOR), as well as tritylium salts. According to the literature, there are very limited examples describing the protection of alcohols as trityl ethers with triphenylmethanol (TrOH) in the presence of an acidic catalyst. Some examples are aqueous H2SO4 [1], B(C6F5)3 [2], ZnCl2 [2], AlCl3 [2] (all in dichloromethane), and Fe(III) salts in alcoholic solvents [3]. When trityl ethers (TrOR, R = benzyl-, p-methoxybenzyl- and prenyl-) were used, protection was carried out in the presence of an oxidizing agent such as 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) [4] or DDQ/Mn(OAc)3 [5]. Triphenylmethyl chloride and bromide (X = Cl, Br) were usually used in the presence of an organic base, such as pyridine [6–8], dimethylaminopyridine (DMAP) [9], 2,4,6-tri-t-butyl pyridine [10], 2,4,6-collidine,[11] triethylamine (TEA) [12,13], and 1,8-diazabicyclo[5.4.0]-undec-7-ene (DBU) [14]. In addition, some tritylating reagents with ionic character such as TrClO4 [8], TrPF6 [11], TrBF4 [15,16], and trityl triflate [17,18] were used alone or in the presence of substituted pyridines. Some of these methods require harsh reaction conditions such as azeotropic removal of water at high reaction temperatures, long reaction times, use of large quantities of organic solvents and bases, and tedious work-up procedures. Furthermore, most of these catalysts are not recoverable, and cannot be used in further subsequent reactions. To the best of our knowledge, there are no reports in the literature on solvent-free protection of alcohols using TrOH, so far.
In recent years, mesoporous materials such as MCMs have received much attention as potential heterogeneous catalysts to replace homogeneous catalytic systems, and have proven to be excellent host materials for developing organic reactions, because of their high surface area, large pore volume, uniform porosity, and excellent thermal-, hydrothermal-, chemical-, and mechanical stability.
In continuation of our previous work on the ability of the nanoporous MCM-41-SO3H catalyst to promote the protection of carbonyls as 1,1-diacylals [19], we herein wish to report the protection reaction of alcohols 1 as trityl ethers under solvent-free conditions, and their deprotection at ambient temperature by using MCM-41-SO3H as a recyclable catalyst (Scheme 1).
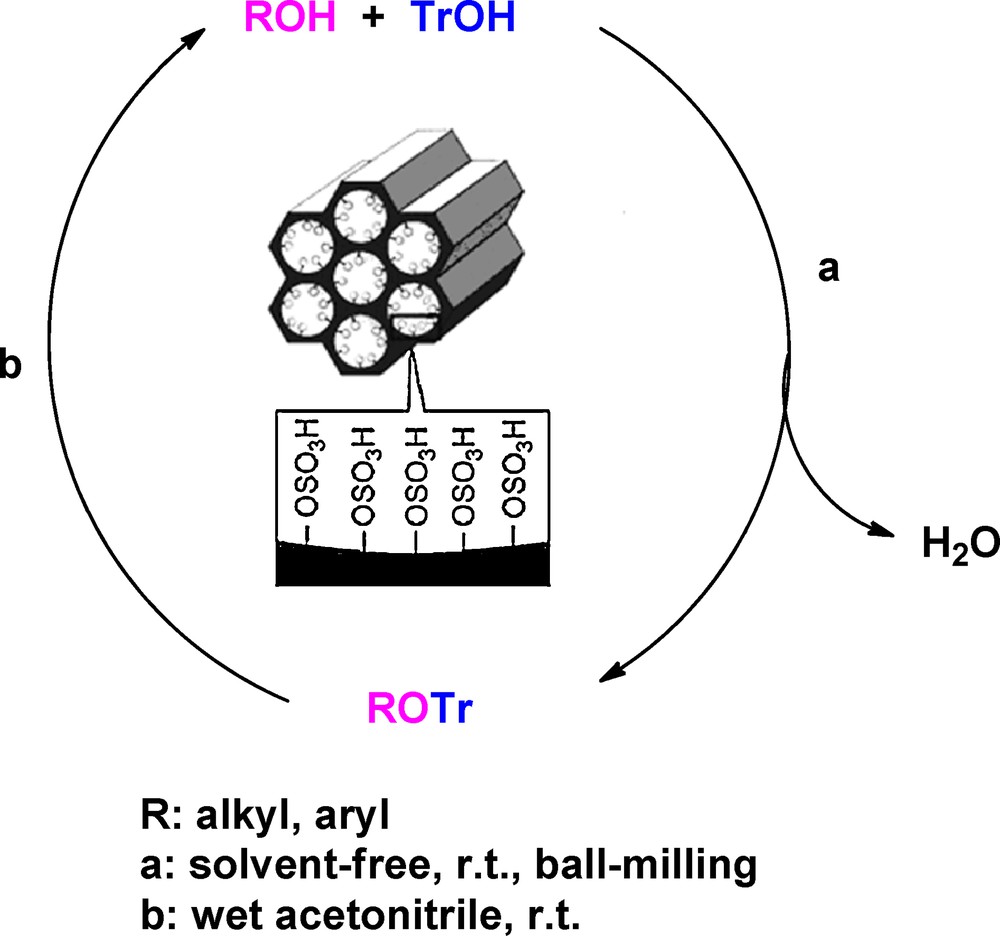
Protection and deprotection reaction of alcohols catalyzed by MCM-41-SO3H.
2 Results and discussion
In order to find the best conditions for the protection reaction of alcohols, the reaction of 1.0 mmol of benzyl alcohol (1a) with 1.0 mmol of triphenylmethanol (2a) by ball-milling at room temperature was chosen as a model reaction. The effect of the catalyst type and of loading was then examined. The catalytic activity of MCM-41 itself and its modified forms such as boron (B-MCM-41), ferric (Fe-MCM-41), aluminum (Al-MCM-41), zirconium (Zr-MCM-41), and MCM-41-SO3H has been studied. The results have been summarized in Table 1.
Protection reaction of benzyl alcohol (1a) with triphenylmethanol (2a) by some modified MCM-41 catalystsa.
| Entry | Catalyst | Amount of catalyst (mg) | Time (min) | Yieldb (%) |
| 1 | – | – | 24 h | 0 |
| 2 | MCM-41 | 15 | 60 | 0 |
| 3 | B-MCM-41 | 15 | 90 | 64% |
| 4 | Al-MCM-41 | 15 | 45 | 59% |
| 5 | Zr-MCM-41 | 15 | 120 | 35% |
| 6 | Fe-MCM-41 | 15 | 60 | 78% |
| 7 | MCM-41-SO3H | 5 | 20 | 59% |
| 8 | MCM-41-SO3H | 10 | 20 | 87% |
| 9 | MCM-41-SO3H | 15 | 20 | 98% |
| 10 | MCM-41-SO3H | 20 | 20 | 96% |
| 11 | MCM-41-SO3H | 100 | 20 | 52% |
a Reaction conditions: benzyl alcohol (1a) (1.0 mmol), triphenylmethanol (2a) (1.0 mmol), catalyst, solvent-free, ball-milling, room temperature.
b Isolated yield.
It is noteworthy to mention that no reaction occurred in the absence of a catalyst, even after 24 h (Table 1, entry 1). As it is seen from the results, MCM-41-SO3H catalyst shows the highest activity, presumably because of its considerable acidic nature. The acidic capacity of MCM-41-SO3H catalyst was determined as 0.011 mmol [H+] g−1 via titration of 1.0 g of the catalyst with a 0.01 N solution of NaOH. This will guarantee much milder reaction conditions compared to aqueous H2SO4 which was previously used [1]. The average pore width (4V/A) of nanoporous MCM-41-SO3H was measured as 2.62 nm by the BJH adsorption method.
By using 5 and 10 mg of MCM-41-SO3H catalyst, moderate yields of 59% and 87% have been obtained after 20 min of ball-milling at room temperature, whereas increasing the loading to 15 mg has practically converted all the alcohol into its corresponding ether (Table 1, entries 6–8). Increasing the amount of the catalyst to 100 mg reduced the yield considerably (Table 1, entry 10).
By employing the optimized reaction conditions, various aromatic and aliphatic alcohols have been subjected to trityl protection. Table 2 shows the scope of the reaction. As it is shown, primary and secondary aliphatic and benzylic alcohols were converted into their corresponding trityl ethers very easily, in high yields and short reaction times.
Tritylation of various alcohols using MCM-41-SO3H under optimal conditionsa.
| Entry | Substrates | Product | Time (min) | Yieldb (%) | Mp (Obsd) (°C) | Mp (Lit.) (°C) |
| 1 | 1a | 3a | 20 | 98 | 103–105 | 102–104 [20] |
| 2 | 1b | 3b | 10 | 98 | 117–118 | 117–117.5 [21] |
| 3 | 1c | 3c | 15 | 96 | 101–102 | Present work |
| 4 | 1d | 3d | 30 | 98 | 136–138 | 137–138 [22] |
| 5 | 1e | 3e | 40 | 97 | 147–149 | Present work |
| 6 | 1f | 3f | 60 | 98 | 168–169 | 166–167 [21] |
| 7 | 1g | 3g | 90 | 96 | 150–151 | Present work |
| 8 | 1h | 3h | 75 | 89 | 118–120 | 118–119 [23] |
| 9 | 1i | 3i | 120 | 61 | 153–154 | 154–155 [12] |
| 10 | 1j | 3j | 90 | 86 | 84–85 | 85 [24] |
| 11 | 1k | 3k | 60 | 94 | 87–88 | 87.1–87.8 [25] |
| 12 | 1l | 3l | 75 | 98 | 124–126 | Present work |
| 13 | 1m | 3m | 105 | 88 | 55–56 | 54–55 [1] |
| 14 | 1n | 3nc | 120 | 95 | 186–188 | 187–188 [26] |
| 15 | 1o | 3o | 180 | – | – | – |
a Reaction conditions: alcohol (1.0 mmol), triphenylmethanol (2a) (1.0 mmol), MCM-41-SO3H catalyst (15 mg), solvent-free, ball-milling, room temperature.
b Isolated yield.
c In the presence of two equivalents of TrOH.
Comparison between the obtained results showed that benzyl alcohols including electron-donating groups such as CH3 (Table 2, entries 2 and 3) required shorter reaction times compared to benzyl alcohols bearing electron-withdrawing groups such as Cl and NO2 (Table 2, entries 4–7). Clearly, the presence of electron-donating groups, especially in ortho and para positions, facilitates the nucleophilic attack of the alcohols. The secondary alcohols 1h–1j reacted slowly at room temperature to yield the corresponding trityl ethers, however affording good yields (Table 2, entries 8–10).
Aliphatic alcohols (Table 2, entries 10–14) need more time compared to aromatic alcohols. In the case of ethylene glycol, both hydroxyl groups were protected by using 2 equivalents of triphenylmethanol. By using one equivalent of TrOH, a mixture of unreacted ethylene glycol along with mono- and deprotected products was afforded.
The protection reaction seems to be sensible to the steric situation of the starting alcohol. Comparing entries 1, 9, and 15, one should conclude that with increasing the steric hindrance at the α-C, the activity of the hydroxyl group would decrease dramatically. It is noteworthy to mention that in the case of tertiary alcohol 1o (Table 2, entry 15), no protection reaction was observed at all. Moreover, in contrast to some previously reported procedures [3], we have not observed any elimination or oxidation products.
According to these facts, a possible mechanism for the catalyzed protection of alcohols by MCM-41-SO3H is shown in Scheme 2. As shown, triphenylmethanol (2) will be protonated at the oxygen atom by the Brønsted acid MCM-41-SO3H. Intermediate I will then dissociate into water and trityl carbocation (II). Nucleophilic attack of alcohol (1) by the trityl cation followed by deprotonation affords the desired product (3).
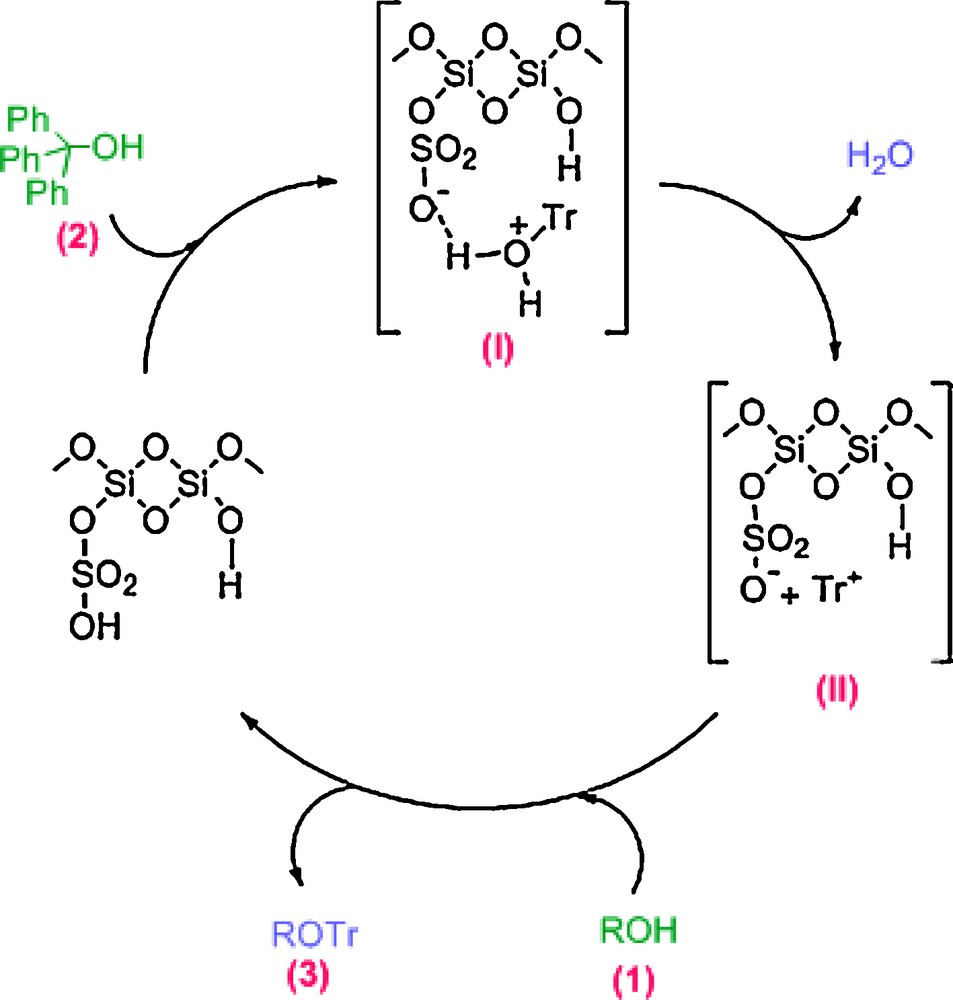
Proposed mechanism for the protection reaction of alcohols with triphenylmethanol catalyzed by MCM-41-SO3H.
It is well understood that trityl carbocation is more stable than benzylic carbocation and can be formed faster. To confirm this in the present method, 0.5 mmol of triphenylmethanol and 0.5 mmol of 4-chlorobenzyl alcohol have been poured in two mortars separately, and MCM-41-SO3H has been added to them. Upon grinding of triphenylmethanol with the catalyst, the color has been changed from white to yellow, which suggests the formation of trityl carbocation. In the mortar containing 4-chlorobenzyl alcohol, no color change was observed and the alcohol remained unchanged as a white powder. The formation of the trityl carbocation has been also approved by UV–Vis spectroscopy (Fig. 1). Thus triphenylmethanol (A), MCM-41-SO3H catalyst (B), and a grinded mixture of triphenylmethanol with MCM-41-SO3H catalyst (C) have been studied using solid-state UV–Visible spectroscopy. As shown in Fig. 1, a new peak at about 440 nm was observed in the C sample spectrum, which is related to the yellow color of Tr+, confirming the formation of the carbocation.
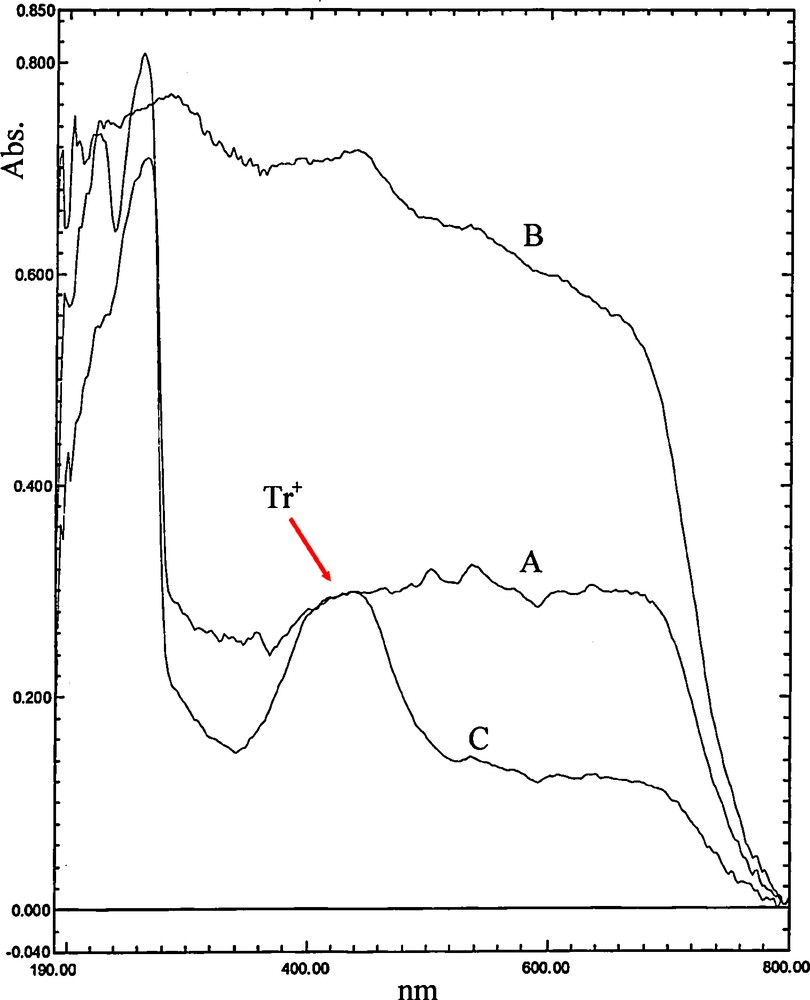
Solid-state UV–Visible spectra of grinded samples of triphenylmethanol (A), MCM-41-SO3H catalyst (B), and a mixture of triphenylmethanol with MCM-41-SO3H catalyst (C).
Interestingly, we have observed that MCM-41-SO3H was also able to catalyze the reverse deprotection reaction into their parent alcohols. In order to obtain the best reaction conditions for this purpose, the reaction of benzyl trityl ether in the presence of MCM-41-SO3H at room temperature has been chosen as a model one and the effect of various solvents on it has been examined. As shown in Table 3, MCM-41-SO3H in wet CH3CN has quantitatively converted benzyl trityl ether to its parent alcohols. In water and methanol, the starting material was sparingly soluble, hence remained practically unreacted. Further investigations show that the deprotection reaction in wet CH3CN will be completed at room temperature in 20 min.
Effect of different solvents on the deprotection of benzyl trityl ether in the presence of a catalytic amount of MCM-41-SO3H at room temperaturea.
| Entry | Solvent | Time (min) | Yieldb (%) |
| 1 | H2O | 120 | - |
| 2 | CH3OH | 120 | 10 |
| 3 | CH2Cl2 | 60 | 56 |
| 4 | CH3NO2 | 90 | 79 |
| 5 | CH3CN (wet) | 20 | 100 |
a Reaction conditions: benzyl trityl ether (20 mg), MCM-41-SO3H (20 mg), solvent (2.5 mL), room temperature.
b Yields were determined by high-performance liquid chromatography (HPLC).
Other primary and secondary, and aliphatic or benzylic trityl ethers were also converted very easily into their corresponding hydroxyl compounds with a catalytic amount of MCM-41-SO3H in wet CH3CN, and in high yields (Table 4).
Deprotection of trityl ethers in the presence of MCM-41-SO3H in CH3CN at room temperaturea.
| Entry | Substrate | Product | Time (min) | Yieldb (%) |
| 1 | 3a | 1a | 20 | 100 |
| 2 | 3b | 1b | 10 | 100 |
| 3 | 3c | 1c | 15 | 100 |
| 4 | 3d | 1d | 20 | 100 |
| 5 | 3e | 1e | 30 | 100 |
| 6 | 3f | 1f | 40 | 100 |
| 7 | 3g | 1g | 45 | 100 |
| 8 | 3h | 1h | 60 | 100 |
| 9 | 3j | 1j | 90 | 100 |
| 10 | 3l | 1l | 40 | 100 |
| 11 | 3m | 1m | 30 | 100 |
| 12 | 3n | 1n | 20 | 100 |
a Reaction conditions: trityl ether (20 mg), MCM-41-SO3H (20 mg), acetonitrile (2.5 mL), room temperature.
b Yields were determined by HPLC and/or GC.
Comparison of the results shows that the substitution on the benzene ring affects the deprotection reaction time. Ethers bearing electron-donating groups such as CH3 on their benzyl moiety (Table 4, entries 2 and 3) reacted in shorter reaction times compared to those bearing electron-withdrawing groups such as Cl and NO2 (Table 4, entries 4–7). Aliphatic trityl ethers were reacted in moderate reaction times with high yields (Table 4, entries 10–12). Trityl ethers with sterically hindered groups preceded the deprotection reaction in longer times (Table 4, entries 8 and 9).
Based on the hard-soft acids and bases rule (HSAB), the acidic proton of MCM-41-SO3H catalyst protonates the trityl ether oxygen, makes it positive and thereby the intermediate III is formed. In the next step, this intermediate dissociates into the trityl carbocation (4) and the alcohol 1. Then, in the presence of wet acetonitrile, 4 converts to triphenylmethanol (2) and the catalyst returns to the catalytic cycle (Scheme 3).
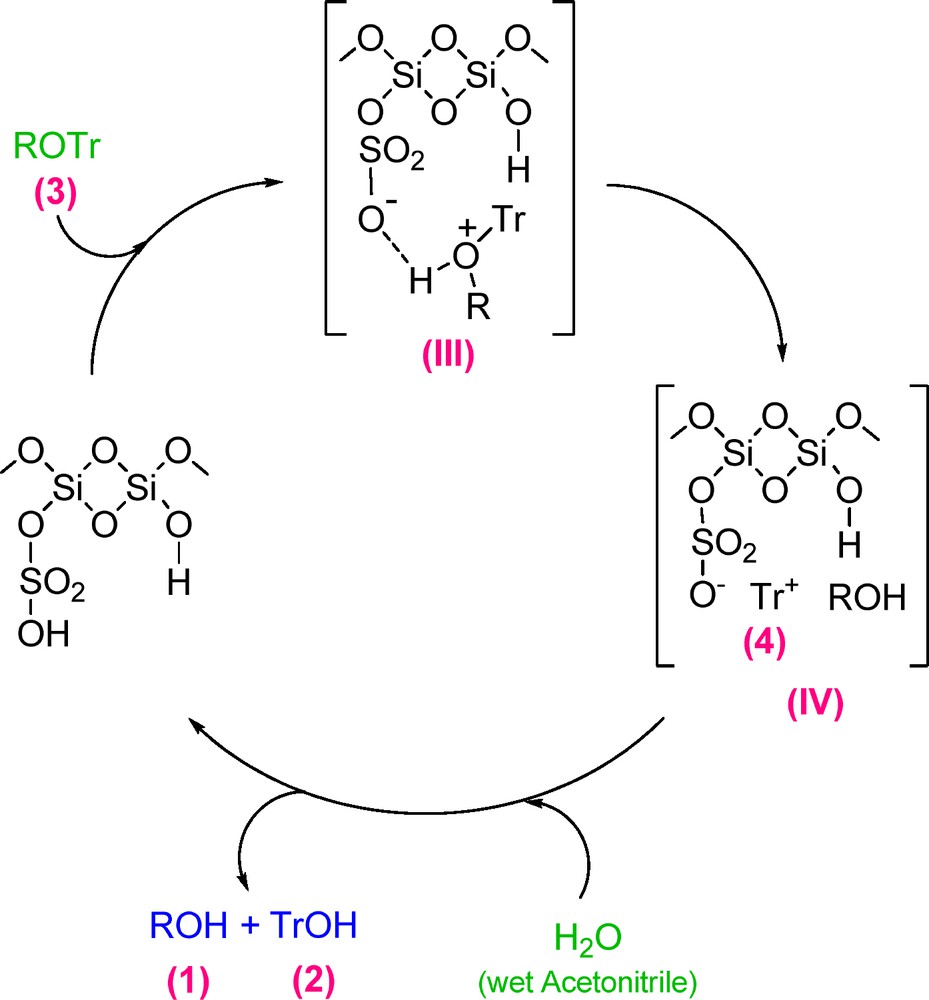
Proposed mechanism for the deprotection reaction of trityl ethers catalyzed by MCM-41-SO3H.
3 Reusability of MCM-41-SO3H catalyst
One of the benefits of heterogeneous catalysts is their easy separation from a reaction mixture and their reusability. The recyclability of the MCM-41-SO3H catalyst has been investigated in the protection reaction of benzyl alcohol 1a with triphenylmethanol 2a. The reactions were carried out according to the above general procedure for the synthesis of trityl ethers. After the first run, the catalyst was washed with EtOAc, dried at room temperature, and then subjected to a second protection reaction. This procedure was repeated three more times (Fig. 2). The average reaction yield for four repeated runs was 95.5%.
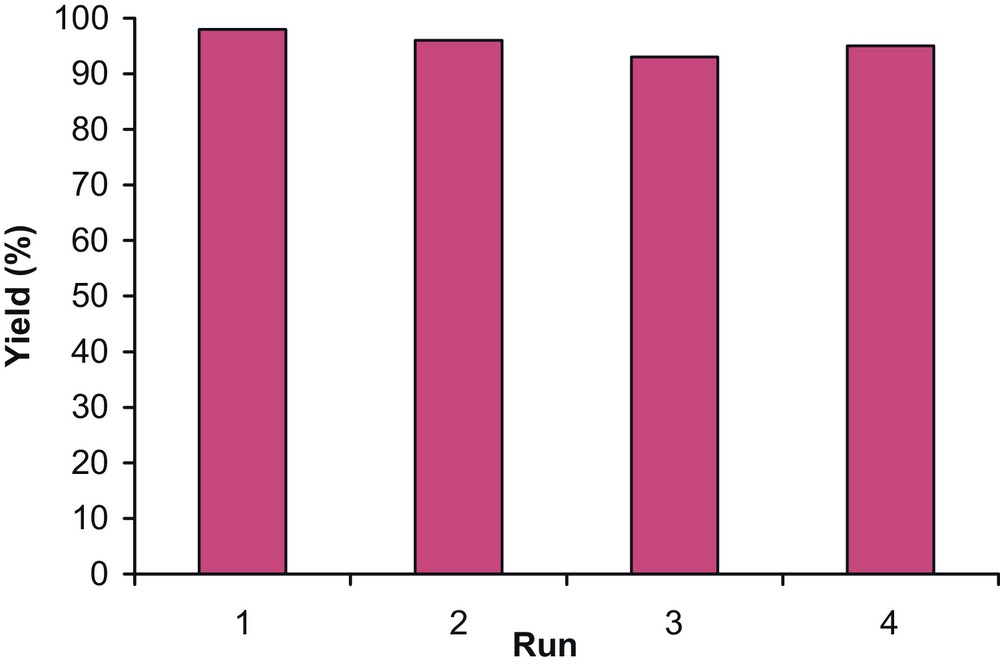
Recyclability of MCM-41-SO3H catalyst for the protection reaction of alcohols.
As shown in Fig. 2, it is possible that some unreacted starting materials or even the product be adsorbed on the surface or in the pores of the catalyst. This part of the materials can be incorporated in the next run (compare runs 3 and 4). This can be principally an advantage of the method, when the catalyst is used industrially in the same reaction.
4 Conclusion
In summary, we have developed a mild, efficient and green procedure for the protection of alcohols as trityl ethers and deprotection of them using the recyclable MCM-41-SO3H catalyst. Moreover, high reaction yields, simple work-up, mild conditions, safe and environmentally benign method, short reaction times, and reusability of the catalyst will make the present method a valuable methodology for the protection and deprotection reaction of alcohols. No by-products (e.g., alkenes from elimination, carbonyls from oxidation, or ethers from homocoupling reactions) were detected. In addition, this catalyst offers the opportunity to protect alcohols as trityl ethers in the presence of a base-sensitive functionality.
Both protection and deprotection reactions proceed with high yields at room temperature in the presence of MCM-41-SO3H. It is noteworthy to mention that attempts at deprotecting the trityl group in similar methods were usually unsuccessful (for example, the deprotection of the trityl group by [B(C6F5)3], even at refluxing temperatures [2]).
5 Experimental
5.1 General
All chemicals and reagents were purchased from Merck, Aldrich, and Fluka and used without further purification. Tetraethyl orthosilicate (TEOS) and cetyltrimethylammonium bromide (CTAB) were used as sources of silicon and structure-directing agent, respectively. Analytical TLC was carried out using Merck 0.2-mm silica gel 60 F-254 Al-plates. MCM-41 was synthesized according to procedures in the literature [27]. MCM-41-SO3H was synthesized according to the reported procedure and characterized by FTIR spectroscopy and BJH analysis [28]. B-MCM-41, Fe-MCM-41 [29a], Zr-MCM-41 [29b], and Al-MCM-41 [30] catalysts were synthesized via literature reports. Known products were characterized by comparison of their melting points and/or spectral data (IR, 1H NMR, and 13C NMR spectra). FTIR spectra were recorded as KBr pellets on a Shimadzu FT IR-8400S spectrometer. 1H NMR (500 MHz) and 13C NMR (125 MHz) spectra were obtained using Bruker DRX-500 Avance and Bruker DRX-300 Avance spectrometers at ambient temperature, respectively. Melting points were determined using an Electrothermal 9100 apparatus and are uncorrected. High-performance liquid chromatography (HPLC, Younglin 9120 model) and gas chromatography (GC, OMEGA WAX 250 model) were used to determine the completeness of the deprotection reactions. A ball mill apparatus (Retsch MM2000 model) having a 20-mL iron cell and two iron balls of diameter 12 mm was used at a frequency of 20 Hz for protection reactions.
5.2 General procedure for the preparation of MCM-41
Diethylamine (2.7 g) was added to deionized water (42 mL) in a 200-mL beaker, while the mixture was stirred at room temperature. After 10 min, CTAB (1.47 g) was added step by step to the above solution under stirring for 30 min, until a clear solution was obtained. Then, TEOS (2.1 g) was added dropwise to the solution. The pH of the reaction mixture was adjusted to 8.5 by slow addition of a 1 M hydrochloric acid solution. At this stage, a precipitate was formed. After 2 h, the solid product was filtered from the mother liquor and washed with deionized water. The sample was dried at 45 °C for 12 h. The synthesized MCM-41 was calcined at 550 °C for 5 h to remove all of the surfactant [27].
5.3 General procedure for preparation of MCM-41-SO3H
MCM-41was modified using a 100-mL suction flask equipped with a constant pressure dropping funnel containing chlorosulfonic acid (ClSO3H) and a gas inlet tube for conducting HCl gas over an adsorbing solution. MCM-41 (1 g) suspended in CH2Cl2 (5 mL) was charged to the flask and ClSO3H (2 mL) was then added dropwise over a period of 30 min at room temperature. HCl gas evolved from the reaction vessel immediately. After complete addition of ClSO3H, the mixture was stirred for 30 min and the solvent was evaporated under reduced pressure to obtain a light gray solid (MCM-41-SO3H) [29]. The MCM-41-SO3H was characterized by BJH analysis and FT–IR spectroscopy. In the FT–IR spectrum, the broad band in the region of 3200–3400 cm−1 is assigned to the O–H stretching vibration of hydroxyl groups. The bands at 1286 cm−1 and 1321 cm−1 are due to the symmetric and asymmetric stretching vibrations of the SO bond of the sulfonic acid group. Moreover, a strong band at 1174 cm−1 is assigned to Si–O–Si asymmetric stretching vibrations and a band at 850 cm−1 related to Si–O–Si symmetric stretching vibrations. The N2 absorption–desorption data determined the nanostructure of the pores of the MCM-41-SO3H: 0.0751 cm3/g for BJH adsorption cumulative volume of pores and BJH adsorption average pore diameter (4V/A) of 2.62 nm The peak values of pore size distribution curves were found for pore diameters of 1.7–2.8 nm
5.4 Typical procedure for the synthesis of trityl ethers
A mixture of alcohol (1.0 mmol) and triphenylmethanol (1.0 mmol) and MCM-41-SO3H (15 mg) as a catalyst along with the two iron balls were fed into the horizontal ball mill vessel. The mixture was grinded at room temperature for 10 min up to 2 h, depending on the alcohol used. The reaction progress was monitored by TLC from time to time. After completion of the reaction, the product was taken out from the cell and dissolved in ethanol and filtered. The solvent was evaporated under reduced pressure and the solid crude was recrystallized from ethanol, if necessary.
5.5 Typical procedure for the deprotection of trityl ethers
A trityl ether (20 mg) and MCM-41-SO3H catalyst (20 mg) were added to acetonitrile (2.5 mL) at room temperature. The mixture was stirred for an appropriate time, indicated in Table 4. After completion of the reaction, MCM-41-SO3H catalyst was separated from the mixture via filtration. Then, acetonitrile was evaporated at reduced pressure.
5.6 Spectral data for the selected products
5.6.1 2-Methylbenzyl trityl ether (3c)
White solid, m.p. 101–102 °C; IR (KBr), ν (cm−1): 3057, 1599, 1445, 1076, 702; 1H NMR (500 MHz, CDCl3): δ (ppm) 2.13 (s, 3H), 4.15 (s, 2H), 7.1–7.7 (m, 19H); 13C NMR (125 MHz CDCl3): δ (ppm) 18.7, 64.1, 87.0, 125.9, 127.1, 127.3, 127.6, 127.9, 128.8, 130.0, 136.1, 137.2, 144.2. Anal. calcd for C27H24O: C, 88.97; H, 6.64; found: C, 89.07; H, 6.77.
5.6.2 2-Chlorobenzyl trityl ether (3e)
White solid, m.p. 147–149 °C; IR (KBr), ν (cm−1): 3082, 1597, 1446, 1369, 1092, 704; 1H NMR (500 MHz, CDCl3): δ (ppm) 4.31 (s, 2H) ppm, 7.2–7.9 (m, 19H); 13C NMR (125 MHz CDCl3): δ (ppm) 63.2, 87.3, 126.8, 127.2, 128.0, 128.1, 128.2, 128.8, 129.0, 132.2, 137.0, 144.0. Anal. calcd for C26H21ClO: C, 81.13; H, 5.50; found: C, 80.61; H, 5.52
5.6.3 2-Nitrobenzyl trityl ether (3g)
White solid, m.p. 150–151 °C; IR (KBr), ν (cm−1): 3082, 3028, 1604, 1520, 1338, 1090, 1067; 1H NMR (500 MHz, CDCl3): δ (ppm) 4.66 (s, 2H), 7.2–8.3 (m, 19H); 13C NMR (125 MHz CDCl3): δ (ppm) 63.1, 87.7, 124.6, 127.3, 127.6, 128.0, 128.5, 128.7, 133.8, 136.0, 143.7, 146.9. Anal. calcd for C26H21NO3: C, 78.97; H, 5.35; N, 3.54; found: C, 78.97; H, 5.39; N, 3.68.
5.6.4 (2-(4-Nitrophenyl)ethyl) trityl ether (3l)
White solid, m.p. 124–126 °C; IR (KBr), ν (cm−1): 3082, 3026, 1601, 1518, 1346, 1078, 703; 1H NMR (500 MHz, DMSO): δ (ppm) 2.94 (t, 2H), 3.18 (t, 2H), 7.22 (m, 15H), 7.46 (AA’BB’, 2H), 8.12 (AA’BB’, 2H); 13C NMR (125 MHz CDCl3): δ (ppm) 37.0, 64.3, 87.2, 123.9, 127.5, 128.3, 129.0, 130.4, 144.3, 147.0, 148.0 ppm Anal. calcd for C27H23NO3: C, 79.20; H, 5.66; N, 3.42; found: C, 79.04; H, 5.72; N, 3.58.
Acknowledgements
We are grateful for the financial support from The Research Council of Iran University of Science and Technology (IUST), Tehran, Iran. Our special thanks go to Dr. M. G. Dekamin for providing us with some M-MCM41s.