

1 Introduction
The ability of bipyridinium salts, best known as viologens, to form electron-responsive π-stacked CT complexes with a variety of conjugated donors, including tetrathiafulvalene or dimethoxynaphtalene, has already been widely exploited to trigger molecular motions within mechanically-interlocked molecular architectures or to develop redox-responsive supramolecular assemblies [1–5]. Further interest for viologen derivatives in supramolecular chemistry arises from their ability to form non-covalent π-dimers in their reduced states [6–9]. Taking advantage of this reversible redox-responsive dimerization process, we have recently reported a series of switchable architectures undergoing intramolecular rotating or tweezer-like motions including preliminary results obtained with viologen-appended calixarenes revealing that the spectroscopic signature of the intramolecular dimers evolves substantially with the extent of the orbital overlap in the π-dimerized species [10–13]. We now wish to report a detailed analysis of the dimerization process occurring between two alkylated bipyridinium radical-cations covalently linked through a flexible calixarene platform [14,15], including experimental and theoretical investigations aiming at providing deeper insights into the redox-triggered association process.
2 Results and discussion
2.1 Synthesis
The synthetic route leading to the targeted calixarene-bipyridinium derivative 44+ is summarized on Scheme 1. The synthesis of 44+ has been reported in a previous article [13]. The first step involves a regioselective O-substitution of the p-tert-butylcalix [4] arene 1 with an excess of 1,2-dibromoethane in the presence of a mild base. The intermediate product 2 is then reacted with an excess of 1-methyl-4-(4′-pyridyl)pyridinium (3) to afford the targeted compounds 44+. Recrystallization of the latter species using a saturated KPF6 aqueous solution afforded 4(PF6)4 isolated in 23% yield.

Synthesis of the calixarene–bipyridinium 44+.
The partial 1H NMR spectra of 44+, recorded in deuterated acetonitrile, is depicted on Fig. 1. The exact attribution of these NMR data has been achieved using 2D NMR spectroscopy. The cone conformation of the calixarene skeleton is supported by the observation of two doublets, at ca. 3.5 and 4 ppm, attributed to the non-equivalent bridging methylenic protons Ha and Hb (Fig. 1). The flexible alkyl chain introduced between the phenol and pyridinium centers appears in the 1H NMR spectrum as two triplets, Hc and Hd, the most deshielded one being attributed to the methylene group linked to the positively charged nitrogen atom. In the aromatic domain, the aryl protons He and Hf are observed as two singlets resonating at ca. 7.2 ppm.
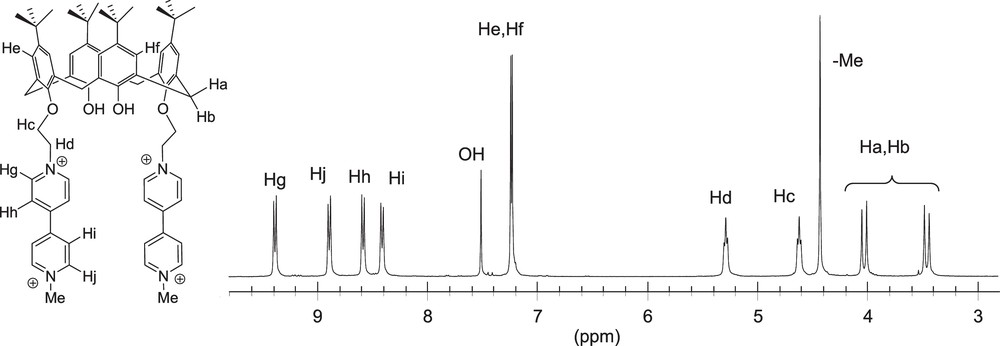
1H NMR spectrum of 4(PF6)4 recorded from 3.0 to 9.8 ppm (CD3CN, 300 MHz).
2.2 Electrochemistry
The π-dimerization of organic radicals has already been observed with a wide range of polyaromatics. Most of the time, formation of π-dimers in solution requires large concentrations in radicals, very low experimental temperatures or use of additives, such as the cucurbiturils and cyclodextrins acting as containers allowing the inclusion of intermolecular π-dimer complexes. The non-covalent association of two organic radicals can also be achieved efficiently without additives in standard conditions, i.e. at room temperature and at low concentration, upon linking both partners through an appropriate covalent bridge.
Use of alkyl, ferrocenyl, phenyl or calixarene linkers of different lengths and geometries (RBP2+ ∼ Fc ∼ BP2+R, RBP2+ ∼ (CH2)n ∼ BP2+R, RBP2+ ∼ Ph ∼ BP2+R, RBP2+ ∼ Calix ∼ BP2+R in Scheme 2) has indeed already been shown to promote the intramolecular dimerization of bipyridinium radicals, the efficiency of the process being tuned by the structure and flexibility of the linker [10–13,16–21].

The ability of bipyridinium cation-radicals (BP+•) to form π-dimer complexes in solution can be readily checked by electrochemical measurements [6,10–13,16,19,22,23]. Key experimental evidences supporting the existence of intra- or intermolecular dimerization processes following the electron transfer are indeed:
- • a large anodic shift of the first viologen-centered reduction potential as compared to the value measured for a simple BP2+/BP+• redox couple used as a reference, and most importantly;
- • the observation of a first reduction wave featuring a peak to peak potential shift lower than 59 mV, a value which is expected to be measured for one-electron Nernstian electron transfers or for multi-electron Nernstian transfers centered on molecules bearing multiple, fully independent and chemically equivalent, redox centers.
The existence of non-covalent associations between bipyridinium cation-radicals BP+• can be further demonstrated by spectroscopic analyses, through the observation of diagnostic electronic absorption bands in the near-IR region and by silent ESR signatures.
The intramolecular dimerization of both reduced bipyridinium units in [4] (Scheme 3), produced in-situ by two electron electrochemical reduction of 44+, has been postulated in a preliminary report on the ground of spectroelectrochemical data [13].
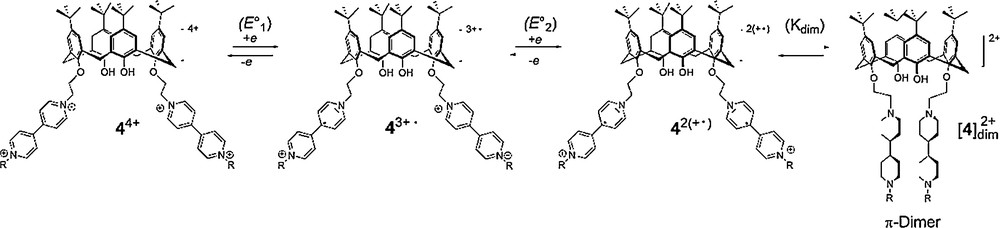
Following these initial findings, we have now carried out a detailed analysis of the electrochemical response obtained with 44+. The voltammetric curves recorded with 44+ in electrolytic ACN solutions exhibit two consecutive bipyridinium-centered cathodic waves observed between −0.5 and −1.5 V, each wave accounting for two electrons (1 electron/bipyridinium fragment) to produce successively 42(+•) and 4. The electrochemical data corresponding to these two consecutive reduction waves are collected in Table 1 along with those of dimethyl-4,4′-bipyridinium (Me2BP2+)24 used as a reference compound.
Peak potentials (Epc and Epa) and ΔEp = Epa − Epc values (mV) measured by cyclic voltammetry for 4(PF6)4 and Me2BP(PF6)2 in ACN (TBAP 0.1 M) at a vitreous carbon electrode (ν = 0.1 V·s−1), E (mV) vs. Ag/Ag+a.
| Epc1 | Epa1 | ΔEp1 | Epc2 | Epa2 | ΔEp2 | |
| Me2BP2+ | −776 | −718 | 58 | −1183 | −1122 | 61 |
| 44+ | −690 | −654 | 36 | −1177 | −1137 | 40 |
a E1/2(ferrocene/ferrocenium) = (Epa + Epc)/2 = +86 mV vs. Ag/Ag+.
The most striking evidence demonstrating the existence of chemical reactions involving the reduced forms of the viologen-appended calixarene (MeBP+• ∼ BPMe+•) comes from comparing the reduction peak potentials measured in the same experimental conditions for 44+ with that of the dimethyl-4,4′-bipyridinium Me2BP2+. The overlaid curves shown on Fig. 2A indeed reveal that the first pyridinium-centered reduction is achieved at a much less negative potential in MeBP2+ ∼ BPMe2+ than in the reference Me2BP2+. This significant shift is obviously not attributed to a different substitution pattern, every BP2+ subunits being quaternized with similar alkyl substituents, but only to the stabilization of the electrochemically generated BP+• radicals in the non-covalent π-dimer complex [4] (Scheme 3).

Electrochemical data for 4(PF6)4 in anhydrous acetonitrile + tetra-nbutylammonium perchlorate (TBAP) 0.1 M, measured by cyclic voltammetry at a stationary vitreous carbon electrode (∅ = 3 mm), E vs. Ag/Ag+ 10−2 M. A. Voltammetric curve of 4(PF6)4 (dotted line), 0.5 mM, compared to that of Me2BP(PF6)2 (full line), 1 mM, ν = 0.1 V·s−1. B. Plot of the peak current values (Ipa, Ipc) measured as a function of ν1/2 (scan rate ν in V·s−1), 0.5 mM. C. Peak potential values (Epa, Epc) plotted as a function of the molar concentration of 4(PF6)4 in DMF + TBAP 0.1 M, (ν = 0.1 V·s−1). D. Plot of the peak potential values (Epa, Epc) and of the peak to peak potential shift (ΔEp) as a function of scan rate (logν), 0.5 mM.
The first reduction wave observed in the CV of 44+, involving the [BP2+]/[BP+•] redox couple (E°1 and E°2 on Scheme 3) and the associated dimerization equilibrium (Kdim on Scheme 3), remains reversible and diffusion-controlled only for scan rates (ν) spanning the range of 0.01 V·s−1 up to around 1 V·s−1, as judged from the negligible effect of the scan rate increase on the peak potential values (Epa and Epc on Fig. 2D), and from the linear variation of the current peaks (Ipa and Ipc) as a function of ν1/2 (Fig. 2B). At higher scan rates, the slope break seen on Fig. 2B suggests a change in the rate limiting step which might as well involve the kinetics of the electron transfer and/or the kinetics of the coupled chemical reactions.
Insights into the thermodynamics and kinetics of the chemical reactions coupled to the electron transfers, i.e. the dimerization and the associated disproportionation equilibriums (2BP+• = BP2+ + BP0), have also been provided by simple electrochemical analysis. The intramolecular character of the dimerization process following the diffusion-controlled electrochemical reduction of 44+ is for instance supported by the fact that the anodic and cathodic peak potentials do not vary over a large concentration range (Fig. 2C).
The relative importance of the kinetics of the chemical vs. electrochemical steps could then be roughly estimated upon checking how experimental data such as the peak potential values and the peak to peak potential shift (ΔEP = Epa − Epc) evolve with the time scale (scan rate) of the electrochemical experiment. The kinetics of a simple electron transfer centered on a bipyridinium center was first checked upon studying the reference compound Me2BP2+. As already seen with 44+, the first reduction of Me2BP2+ remains Nernstian only from scan rates ranging from 0.05 V·s−1 up to around 1 V·s−1, the cathodic and anodic peak potentials being then progressively shifted as the rate is further increased. In the experimentally accessible time scale, the concomitant increase in the associated ΔEp value, found to range from 60 mV, measured at 0.05 V·s−1, up to 75 mV measured at around 10 V·s−1, suggests that the system gets gradually under kinetics control, i.e. limited by the rate of the heterogeneous electron transfer.
Similar investigations have then been carried out with the bis-viologen calixarene derivative 44+. The plot depicted on Fig. 2D shows that the ΔEp value is also submitted to quite significant changes, while remaining at all scan rates below the standard 59 mV value expected for simple reversible redox systems, from 38 mV measured at 0.01 V·s−1 up to 54 mV, measured at almost 10 V·s−1. In our experimental conditions and in the investigated time scale, the good correlation found between the electrochemical behavior of 44+ and Me2BP2+ supports the fact that the dimerization of the bipyridinium radicals in 42(+•) proceeds with a fast kinetics and that the rate limiting process is the heterogeneous electron transfer.
2.3 Computational chemistry
The energetic gain due to the π-dimerization is a fundamental thermodynamic parameter, which can be determined by computational chemistry. The non-covalent association of two bipyridinium cation-radicals can be described as a delicate balance between charge repulsion and VDW attractive dispersion forces due to the formation of a “π bond” between two BP+• centers. An intuitive description of such interaction was first provided by non-covalent interactions (NCI) [25,26] calculations conducted on the calixarene π-dimer [4] . The side view shown on Fig. 3 especially brings to light the importance of the dispersive contribution, represented as a continuous surface located between both bipyridinium units, and of the repulsive electrostatic contribution, seen as spheres in the center of each pyridinium rings (Supplementary data, A colored version of this figure is available in the ESI section).

Side view of [42+]dim modeled by non-covalent interactions calculations (Supplementary data, see the electronic supporting information for a more detailed analysis) [25,26].
We have already reported a methodological study demonstrating the relevance of DFT and DFT-D methods to the description of such weak intra-molecular interactions [10]. This previous work involved different calculation procedures such as the most popular MP2, B3LYP and BLYP models as well as the earliest DFT-D method (CAM-B3LYP and B97D). Following these investigations, we report in the present article calculations, which have been carried out with the latest DFT-D methods, including the one recently developed by DiLabio [27] to take simultaneously into account basis set superposition error (BSSE) and dispersion corrections.
The calculated stabilization energy associated with the intramolecular π-dimerization of two bipyridinum radical-cations covalently linked through a propyl chain (RBP2+ ∼ (CH2)n ∼ BP2+R [Scheme 2, n = 3, R = CH2CONH2]) [10], a ferrocenyl pivot (RBP2+ ∼ Fc ∼ BP2+R [Scheme 2, X = Ph, R = Me]) and through a calixarenyl skeleton (44+) are collected in Table 2. What can be inferred from these data is that the solvent plays a key role in the stabilization of π-dimers. Use of “polar” solvents featuring strong dielectric constants indeed reduces the charge repulsion forces occurring between both positively charged BP+• units and allows them to get closer to one another. The large solvent effect transpiring from the values reported in Table 2 is moreover in full agreement with the fact that π-dimerization is usually only observed in highly polar solvents such as DMF, water or acetonitrile. We also found that the magnitude of the solvent effect is directly related to the amplitude of the variation in dipolar moment values associated with the dimerization process . The stabilization energy due to solvent effects is indeed seen to range from 20 kcal/mol, calculated with RBP2+ ∼ Fc ∼ BP2+R, whose dimerization comes along with a large increase (∼ 10 Debye) [12] of the calculated dipolar moment, down to only 5 kcal/mol obtained with 44+, whose dimerization leads to a much lower variation of the dipolar moment (∼ 2 Debye calculated at the M06-2X level). Another important result is that the solvent effects are not DFT dependent.
Energy differences between the non-associated (RBP+• ∼ BP+•R) π-dimerized forms computed at various level for 44+, RBP2+ ∼ (CH2)n ∼ BP2+R (Scheme 2, n = 3, R = CH2CONH2) [10], RBP2+ ∼ Fc ∼ BP2+R (Scheme 2, X = Ph, R = Me) [12]. Negative signs indicate that the π-dimerized species is more stable.
| Solvent (ACN) | Dispersion correction | BSSE correction | 44+ | RBP2+∼Fc∼BP2+R | RBP2+∼(CH2)n∼BP2+R | |
| M06-2X/6-31 + G** | x | −9.90 | −15.36b | −25.03 | ||
| PCM-M06-2X | x | x | −23.90 | −34.62 | −29.19 | |
| DCP-B3LYP | x | x | +0.10 | −6.67 | −14.35 | |
| PCM-DCP-B3LYP | x | x | x | −14.46 | −25.51 | −19.31 |
| B3LYP/6-31 + G** | +15.97 | +10.06b | −0.94a |
The calculated values collected in Table 2 also vary to a very large extent with the structure of the covalent linker introduced between both π-dimerizable centers. The largest stabilization energy is obtained with the propyl-linked derivative RBP2+ ∼ (CH2)n ∼ BP2+R (Scheme 2, n = 3, R = CH2CONH2) [10] but we found that the amide substituents, introduced on the terminal nitrogen position, play a major role in the stabilization process. This effect has been revealed upon studying, in the same conditions, the phenyl substituted analog RBP2+ ∼ (CH2)n ∼ BP2+R (Scheme 2, n = 3, R = Ph), which led to a positive energy difference of +5.40 kcal/mol (calculated at the B3LYP/631 + G** level), as opposed to the −0.94 kcal/mol obtained with the amide substituted derivative RBP2+ ∼ (CH2)n ∼ BP2+R. (Scheme 2, n = 3, R = CH2CONH2). The influence of the amide groups was further demonstrated by Symmetry-Adapted Perturbation Theory (SAPT) [28] calculations giving access to the total interaction energy as well its decomposition in physical terms. These calculations were found to yield an interaction energy of around −3.08 kcal/mol with small contributions of the electrostatic and induction terms of around −4.6 kcal/mol and – 2.0 kcal/mol, respectively, the latter being partially quenched by an exchange contribution of +4.5 kcal/mol and an exchange-induction contribution of +2.2 kcal/mol. SAPT calculations also yielded a very large dispersion force contribution of −3.8 kcal/mol, which happens to be much larger than the total interaction energy estimated at 3.08 kcal/mol.
These calculations, and the geometry of the minimized structure, thus lead us to conclude that the amide groups are interacting with one another through dispersion forces, and not through hydrogen bonds as it was initially suspected.
Our calculations moreover suggest that the ferrocenyl-linked π-dimer is slightly less stable than the propyl-linked one and that the least stable π-dimer is the calixarene [4] , this trend being obtained with all the investigated DFT methods. We can also see that, in agreement with previous works [10], B3LYP lacks the dispersion contribution whereas M06-2X seems to overestimate the interaction and that the DiLabio model (DCP-B3LYP) [27], including BSSE and dispersion corrections, appears particularly suited to describe the non-covalent interactions involved in π-dimers.
3 Conclusions
On the ground of advanced electrochemical, spectroscopic and theoretical data, we have demonstrated that two bipyridinium radicals introduced on the lower rim of a calixarene skeleton can be efficiently and reversibly π-dimerized. A detailed electrochemical investigation supports the conclusion that the non-covalent association between both electrogenerated radical-cations proceeds intramolecularly with a fast kinetics, the electrochemical behavior being mostly limited, in the accessible scan range, by the kinetics of the heterogeneous electron transfer. Further insights into the structure and stability of a series of covalently linked π-dimers have been provided by DFT calculations carried out at various computational levels. A major result of these theoretical investigations is that solvents and dispersion-correcting potentials have to be considered to accurately model the non-covalent interactions involved in π-dimer complexes.
4 Experimentals
4.1 Level of calculations
Optimizations of the dimerized structures has been carried out using the Quickstep code of CP2K [29] at the BLYP level using additional corrections from Grimme for dispersion [30] in conjunction with the DZVP basis set for the optimizations of the structures. M06-2X, B3LYP and DCP-B3LYP [27] calculations were performed using the Gaussian suite of codes [31]. Inclusion of solvent effects (acetonitrile) was achieved using the Polarizable Continuum Model [32–35].
4.2 Syntheses
All chemicals were used as received unless described otherwise. 4,4-Dipyridyl (98%) was purchased from Acros chemicals. Anhydrous acetonitrile (ACN) was purchased from Rathburn Inc. 1-Methyl-4-(4′-pyridyl)pyridinium iodide and 1,1′ dimethyl-4,4′-bipyridinium bis(hexafluorophosphate) were synthesized according to literature procedures [24,36].
The synthesis of the brominated derivatives 2 and 3 [37] and of the bipyridinium appended calixarene 4(PF6)4 [13] have been achieved following procedures described in the literature.
4.3 Electrochemistry
Cyclic voltammograms (CV) have been recorded on a ESP300 Biologic instrument. All analytical experiments were conducted under an argon atmosphere (glove box or argon stream) in a standard one-compartment, three-electrode electrochemical cell. Tetra-n-butylammonium perchlorate (TBAP) was used as supporting electrolyte (0.1 M) in non-aqueous media. An automatic Ohmic drop compensation procedure was systematically implemented prior to recording CV data. CH instrument vitreous carbon (ϕ = 3 mm) working electrodes were polished with 1-mm diamond paste before each recording. A CH instrument Ag|AgNO3 (10−2 M + TBAP 0.1 M in ACN) electrode was used as a reference.
Acknowledgments
The present work was supported by the “Agence national de la recherche” (ANR-12-BS07-0014-01), by the “Région Rhône-Alpes” and by the Labex Arcane (ANR-11-LABX-0003-01). The Authors would also like to thank the CECIC for providing access to computing facilities.