1 Introduction
Currently metal oxides have a large range of functional applications and are technologically interesting in many fields: data storage, microelectronic, energy conversions, catalysts, sensors [1–5]. Most of the applications require oxides at the nanometer scale. A large variety of elaboration processes have been developed to produce these metal oxides at the nanoscale as precipitation and decomposition of metal salts, electrochemical methods, reverse micelles as nanoreactors for the oxide formation and sol–gel techniques [6–12]. Another way consists in the use of a hard template, such as the porosity of ordered mesoporous silica for the oxide formation [13–15]. However, in the case of mixed oxide, the classical synthetic methods bring together several precursors of the oxide in solution, making difficult the control of the stoichiometry of the final product.
Whereas PBAs have most often been used as precursors of alloys [16–19], some examples concerning the thermal treatment of PBAs to obtain mixed oxides are reported in the literature: FeCo [20], ZnFe [21], ZnCu [22] or TiFe [23]. Indeed, Prussian blue analogues (PBA) are good candidates for the elaboration of mixed oxides with controlled stoichiometry by thermal decomposition in oxidative atmosphere [22,24–26]. Actually PBA are constituted of a perfect organization of transition metal ions A and B in an ordered network with metal ions homogeneously mixed at the atomic scale. Indeed, the ambidentate cyanide ligands act as asymmetric bridges between the metals A and B, thus forming three-dimensional sequences –A–NC–B–CN–A– [27,28]. A high number of possible combinations of A and B ions originates from the extremely versatile chemistry of these systems. Then, a thermal treatment under oxidative atmosphere should allow the transformation of PBA with perfectly controlled stoichiometry into the corresponding monometallic or mixed oxide by the decomposition of the CN bond. This method is expected to adjust very precisely the chemical composition and the metals organization in the oxide at the molecular level by the control of stoichiometry of the PBA. Hence, this approach could lead to the formation of mixed oxides with a perfect control of the structure and the composition giving particles with adjusted properties.
The physical properties of the functional objects also depend on their size and shape, the control of these parameters and the study of their effect on the properties are a prerequisite for future applications. Several methods have been proposed to confine PBAs particles into mesostructured silica matrix: layer-by-layer growth in the functionalized porosity [19,29], direct reaction between the PBA precursors in the organic phase of the hybrid material [30–32]. We developed another method consisting of two steps:
- • elaboration of a Co2+-containing mesoporous silica monolith;
- • formation of the PBA particles by a simple impregnation step [26,33,34].
This method combines two advantages:
- • the one-step precipitation of the PBA particles;
- • the control of the morphology of the PBA particles by the well-defined porosity.
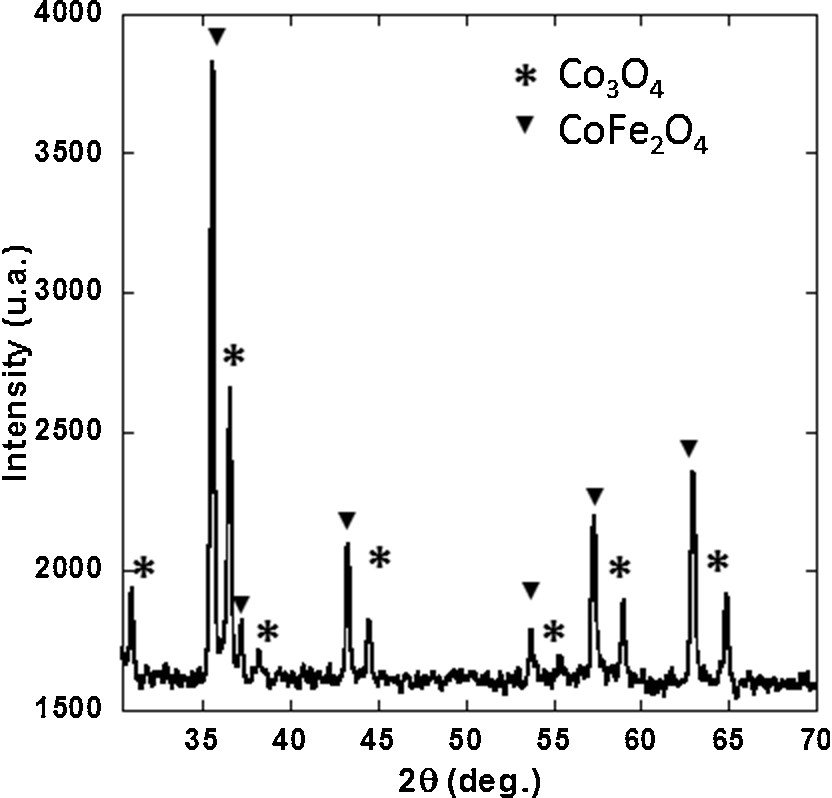
In this preliminary work, a monometallic CoCo PBA precursor was chosen as Co oxide precursor in order to avoid phase demixing that can occur in bimetallic compounds (Supplementary data, Fig. S1). Powdered CoCo PBA freely precipitated in aqueous solution, and CoCo PBA confined into 2D-hexagonal mesoporous silica monoliths were heated under oxidative atmosphere. The products of the thermal treatment were studied and compared.
2 Experimental
2.1 Synthesis of powdered CoCo PBA (CoCo)
The alkali cation-free powdered CoCo PBA was prepared by adding 400 mL of an aqueous solution of potassium hexacyanocobaltate(III) (2.5 mmol L−1, 332 mg) to 100 mL of a pink aqueous solution of Co(II) nitrate (50 mmol L−1, 1.455 g) under vigorous stirring. The addition rate was regulated to last 3 h. The pink powder was centrifuged, washed three times with distilled water, and allowed to dry in air at room temperature. This sample is called CoCo in the following.
The synthesis method for the elaboration of the CoCo PBA/SiO2 nanocomposite has been adapted from a synthesis procedure previously developed by the authors [26,33,34].
2.2 Synthesis of 2D-hexagonal Co2+-containing ordered mesoporous silica monoliths
2D-Hexagonal mesostructured silica monoliths were synthesized using tetramethyl orthosilicate (TMOS) as the silica source and an amphiphilic triblock copolymer poly(ethylene glycol)-block-poly-(propylene glycol)-block-poly(ethylene glycol) with Mn = 5750 (Pluronic P123) as the structuring agent. Then, 2.4 g of P123 were added to 4 g of TMOS (3.9 mL) in a 30-mL polypropylene vial and the solution was stirred in a water-bath at 50 °C until the polymer had completely dissolved. After cooling to room temperature, 2 mL of a pink aqueous acidic cobalt(II) nitrate hexahydrate solution (0.137 mol L−1, 80 mg) with pH = 1.4 were quickly added to the stirred silicate–block copolymer clear solution and stirred during 2 min 30 s. Then, the pink solution was divided in four vials, which were sealed and placed for one hour in a water thermostated bath at 23 °C without stirring. After removing the vial lid, the sol gelled to give after one week, a pink glassy silica-copolymer monolith containing Co2+ ions in octahedral (Oh) symmetry.
In order to liberate the porosity, the monoliths were heated up to 500 °C. During the thermal treatment, the monolith's color changed from pink to blue due to the thermohydrolysis of the Oh Co2+ complexes, giving hydroxylated tetrahedral (Td) species unreactive towards hexacyanocobaltate(III) [34]. Monoliths were stored in an oven at 80 °C.
2.3 Synthesis of CoCo Prussian blue analogue–silica-nanocomposites (Mono CoCo)
The blue monolith was impregnated using the double-solvent method [13,35,36]. The synthesis of the CoCo PBA/SiO2 nanocomposite consists of two steps:
- • the monoliths were immersed in 3 mL of hexane;
- • impregnated with a volume corresponding to 80% of the porous volume of a potassium hexacyanocobaltate(III) aqueous solution (0.1 mol L−1) containing nitric acid (1.13 mol L−1).
In previous works, we showed that all the Co ions initially added to the sol are transformed in PBA [34] and that the PBA particles were entirely confined within the pores of the silica matrix [33]. The impregnation solution was acidified in order to transform the unreactive hydroxylated Co2+ Td species into reactive [Co(H2O)6]2+ PBA precursors. The color of the monolith instantly changed from blue to pink. After 5 min, the monolith was removed from hexane and was washed 3 times with distilled water to eliminate all the unreacted species. It was then allowed to dry at room temperature. This sample is called Mono CoCo in the following.
2.4 Thermal treatment of CoCo and Mono CoCo
In order to transform the CoCo PBA into Co oxide, CoCo and Mono CoCo were heated under air at 700 °C during 2 h. These samples are called CoCo–Ox and Mono CoCo–Ox in the following.
2.5 Materials characterizations
Elemental analyses of Si, Co, C, N, and H were performed at the analysis facility of the CNRS in Solaize.
Thermo differential (DTA) and thermo gravimetric (TGA) analyses were performed under air with a flow rate of 100 mL/min and a temperature ramp of 5 °C/min up to 900 °C using a TA Instruments SDT Q600 apparatus.
Fourier Transform–Infrared (FT–IR) spectra were collected in the transmission mode using a Perkin Elmer Spectrum100 spectrometer. Samples were prepared as KBr pellets (oxides) or nujol suspension (PBA).
X-ray diffraction patterns were recorded using a PAN X’Pert PRO MPD X-ray diffractometer (Cu Kα1).
Transmission electron microscopy (TEM) images were recorded with a JEOL JEM 100CXII electron microscope operating at an acceleration voltage of 100 kV. Silica monoliths were analyzed after ultramicrotomy. For high-resolution microscopy (HRTEM) images, we used a JEOL JEM 2010 apparatus equipped with a LaB6 filament and operating at 200 kV. The images were collected with a 4008 × 2672 pixel CCD camera (Gatan Orius SC1000).
Small-angle X-ray diffraction measurements were performed with a Nanostar laboratory instrument from Bruker-AXS (Karlsruhe, Germany). X-rays (wavelength λ = 1.54 Å) originating from a rotating anode generator Microstar with microfocus (100 × 100 mm2) going through a double multilayer element optics in a Montel arrangement. This set-up yields a slightly divergent monochromatic beam with a circular 1-mm diameter cross-section at the sample level. Ground samples are placed in 1.5 mm diameter capillaries that are inserted in an automatic sample changer with 13 positions. Scattered photons are detected using a bidimensional gas detector (Vantec 2000) positioned at 71 cm from the sample. Direct beam position and sample-detector distance are accurately determined using a reference scatterer, silver behenate, a lamellar compound with a well-characterized interlamellar distance (dl = 58.38 Å). The entire beam path from the exit of the Montel optics to the detector entrance window is evacuated so as to minimize background scattering. Scattering patterns were recorded with exposure times ranging from 10 to 30 min. Two-dimensional images are corrected for detector distortions using a manufacturer-supplied software before radial integration. For each sample, we obtained in this way the scattered intensity I(q) as a function of the momentum transfer q (q = [4πsin(θ)/λ]) over the q-range [0.012 Å−1–0.4 Å−1].
Magnetic measurements were performed in a SQUID magnetometer (Quantum Devices MPMS5). The magnetic susceptibility (χ) was measured in the heating mode over the 2-300 K temperature range using a 1000 Oe magnetic field.
3 Results and discussion
3.1 CoCo PBA and CoCo PBA silica-nanocomposite
Powdered CoCo PBA and CoCo PBA confined in the porosity of ordered mesoporous silica monoliths were used as oxide precursors. The characterization of these compounds is described in this section.
3.1.1 CoCo PBA in powder form (CoCo)
Fig. 1(a) shows the diffractogram of CoCo. The diffractogram exhibits the diffraction lines of the well-known face-centered cubic structure of PBAs. The diffraction lines were indexed within the space group Fm3m. The a-cubic lattice parameter was calculated from the position of the (200), (220), (400) and (420) diffraction lines: a = 10.25 ± 0.02 Å.

(a) X-ray diffraction patterns and (b) IR spectra of CoCo and Mono CoCo.
Fig. 1(b) shows the IR spectrum of CoCo over the 2050 cm−1–2300 cm−1 cyanide vibration range. The spectrum displays one intense band centered at 2173 cm−1, which was attributed to the vibration of CN in the CoIII–CN–CoII environment. The energy position of this band confirms the formation of a CoCo PBA.
The results of the elemental analyses are gathered in Table 1. The chemical formula K0.1CoII4[CoIII(CN)6]2.7·20 H2O was deduced from these analyses.
Microanalysis results and proposed formula for CoCo.
| CoCo | Co | C | N | H | K |
| % Experimental | 32.85 | 17.16 | 19.35 | 3.13 | 0.32 |
| % Calculated | 33.51 | 16.5 | 19.25 | 3.39 | 0.33 |
| Formula | K0.1Co4[Co(CN)6]2.7·20 H2O |
The TEM micrograph of CoCo (Supplementary data, Fig. S2) shows ill-defined particles ranging in size from 100 nm to 160 nm. The CoCo PBA particles very likely underwent some damages under the electron beam.
This study shows that CoCo is a well-crystallized CoCo PBA powder, containing a very small amount of alkali cations of chemical formula K0.1CoII4[CoIII(CN)6]2.7·20 H2O with particles ranging in size from 100 nm to 160 nm.
3.1.2 CoCo PBA silica-nanocomposite (Mono CoCo)
The organization of the porosity of the silica matrix was checked by small-angle X-ray diffraction. Fig. 2 shows the small-angle X-ray powder diffraction pattern of Mono CoCo. The diffractogram exhibits three peaks which were indexed as the (100), (110) and (200) reflections of a 2D-hexagonal organization. The Co2+-containing silica monolith exhibits a 2D-hexagonal arrangement of mesopores [37].
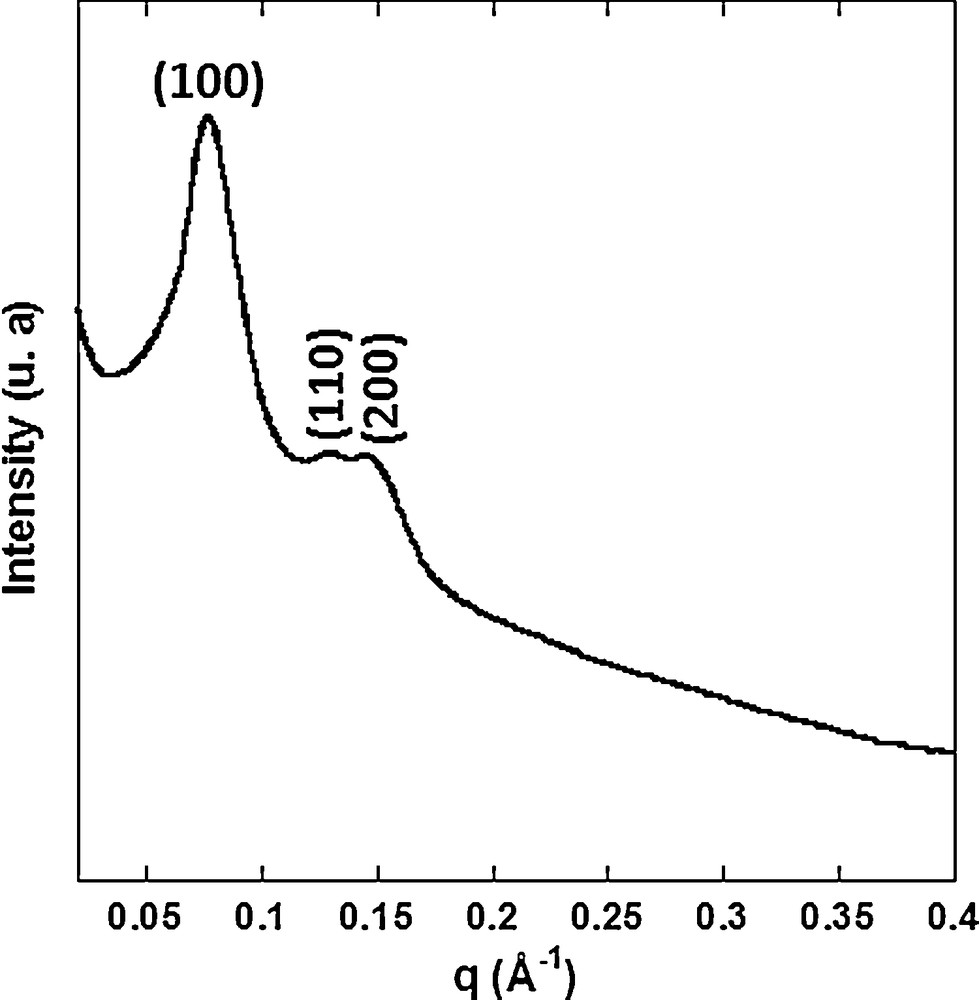
Small-angle X-ray diffraction pattern of Mono CoCo.
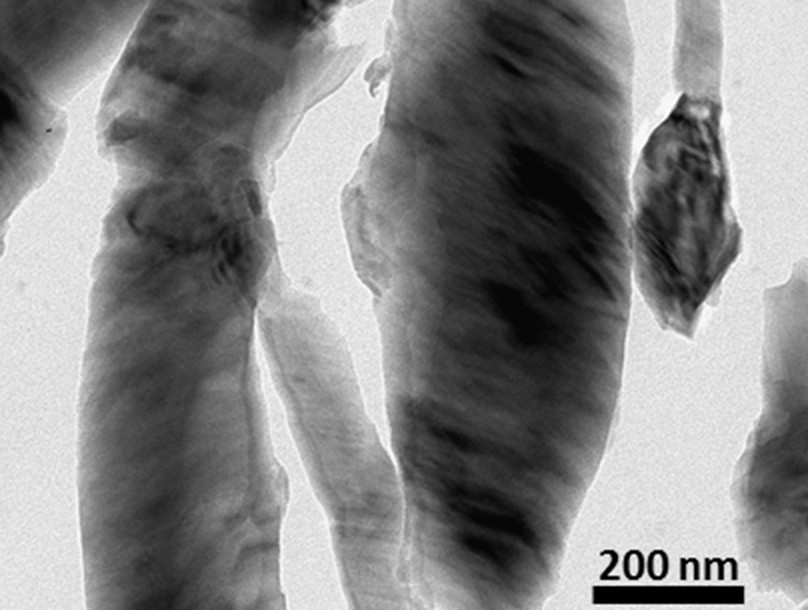
Fig. 3 shows TEM micrographs of Mono CoCo. No CoCo PBA particle was observed outside the monolith in agreement with the full confinement of PBA particles [38]. The images confirm the 2D-hexagonal organization of the porosity. The cylindrical mesopores are 5 nm in diameter. The dark regions indicate the presence of PBA particles in the pores of silica. However, the PBA particles in the porosity are hardly visible because they very likely underwent some damages under the electron beam.

TEM images of Mono CoCo (a) longitudinal section and (b) transversal section.
As alkali cation-containing PBAs are generally more robust in front of an electron beam, a nanocomposite made of such a CoCo PBA was synthesized in exactly the same conditions as those used for the elaboration of the alkali cation-free CoCo PBA/SiO2 nanocomposite, except the presence of an excess of alkali cation in the impregnation solution. A MET image of this compound is shown in Supplementary data, Fig. S3. PBA particles can be seen as dark area in the porosity of this compound.
Fig. 1(a) shows the diffraction pattern of Mono CoCo compared to that of CoCo. The diffraction pattern of Mono CoCo shows the same peaks than the one of CoCo, indicating the presence of CoCo PBA in Mono CoCo. Moreover, the peaks are broader and less intense than those of CoCo, which can be explained by a smaller amount of PBA in the nanocomposite and a smaller PBA particle size.
Fig. 1(b) shows the IR spectrum of Mono CoCo over the 2050 cm−1–2300 cm−1 cyanide vibration range. The spectrum of Mono CoCo shows a band centered at 2177 cm−1 assigned to cyanide in the CoIII–CN–CoII environment. The energy position of this band confirms the formation of a CoCo PBA.
The results of the elemental analyses of Mono CoCo are gathered in Table 2. The chemical formula K0.1CoII4[CoIII(CN)6]2.7(SiO2)489·223 H2O was deduced from these analyses.
Microanalysis results and proposed formula for Mono CoCo.
| Mono CoCo | Co | C | N | Si | H | K |
| % Experimental | 1.06 | 0.5 | 0.74 | 38.72 | 1.26 | < 250 ppm |
| % Calculated | 1.09 | 0.57 | 0.66 | 40.17 | 1.3 | 0.01 |
| Formula | K0.1Co4[Co(CN)6]2.7(SiO2)489·223 H2O |
The whole study shows the formation of CoCo PBA into the ordered porosity of silica monolith.
The powdered CoCo PBA (CoCo) and CoCo PBA confined in the ordered mesoporosity of silica monoliths (Mono CoCo) were used as oxide precursors in the following.
3.2 Thermal treatment of PBA precursors
In order to transform the CoCo PBA into Co oxide, CoCo and Mono CoCo were calcined under air.
Photographs of Mono CoCo before and after calcination are shown in Fig. 4. The monolith is 2 cm in diameter. Its color changes from pink to black after the thermal treatment.
(Color online). Photographs of Mono CoCo (right) and Mono CoCo–Ox (left).
3.2.1 Differential thermo analysis (DTA)/thermo gravimetric analysis (TGA)
TGA and DTA were performed in order to check the effects of the thermal treatment on the compound.
DT and TG analyses of CoCo and Mono CoCo are shown in Fig. 5. Fig. 5(a) shows the thermal analysis of CoCo. A first weight loss of 27.5% from room temperature to 160 °C is accompanied by an endothermic peak on the DTA curve. This step corresponds to the loss of 18 water molecules. The second weight loss of 24% from 280 °C to 320 °C is accompanied by a strong exothermic peak on the DTA curve at 326 °C. This second step corresponds to the decomposition of the cyanide bridges. The XRD pattern of the sample heated up to 900 °C is characteristic of Co3O4 with the spinel crystal structure.
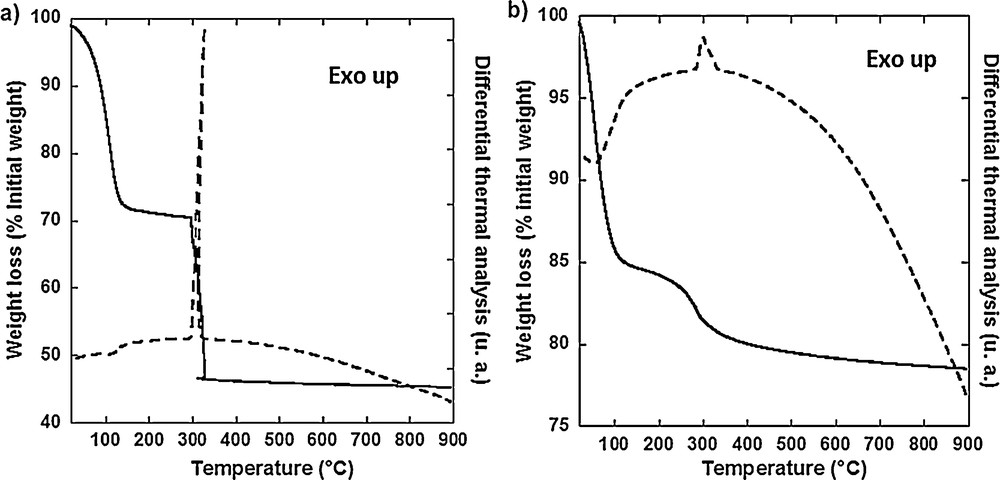
Differential thermal and thermogravimetric analyses of (a) CoCo and (b) Mono CoCo.
Fig. 5(b) shows the thermal analysis of Mono CoCo. The weight loss of 15% from room temperature to 115 °C is accompanied by an endothermic peak on the DTA curve. This weight loss corresponds to the elimination of water filling the silica porosity and water contained in the PBA particles. The second weight loss of 4% from 170 °C to 330 °C is accompanied by an exothermic peak on the DTA curve at 282 °C. This weight loss is due to the decomposition of the cyanide bridges. After this second step, the curve keeps on slowly decreasing due to further condensation of silica.
The DTA and TGA studies show that the cyanide exothermic decomposition occurs 40 °C lower and covers a broader range of temperature for Mono CoCo than for CoCo. These differences can be assigned to confinement and/or to particle size reduction effects in the case of Mono CoCo. This result is in line with a fully confined transformation of CoCo PBA within the porosity of the silica monolith. The temperature of the thermal treatment was chosen above 600 °C in order:
- • to completely decompose the cyanide bridges;
- • to form Co3O4 oxide particles with a high degree of crystallinity.
3.2.2 SAXS
The effect of the thermal treatment on the organization of the silica monoliths was studied by SAXS. The small-angle X-ray powder diffraction patterns of Mono CoCo calcined at 600 °C, 700 °C and 800 °C during 2 h are compared to the one of Mono CoCo in Fig. 6.
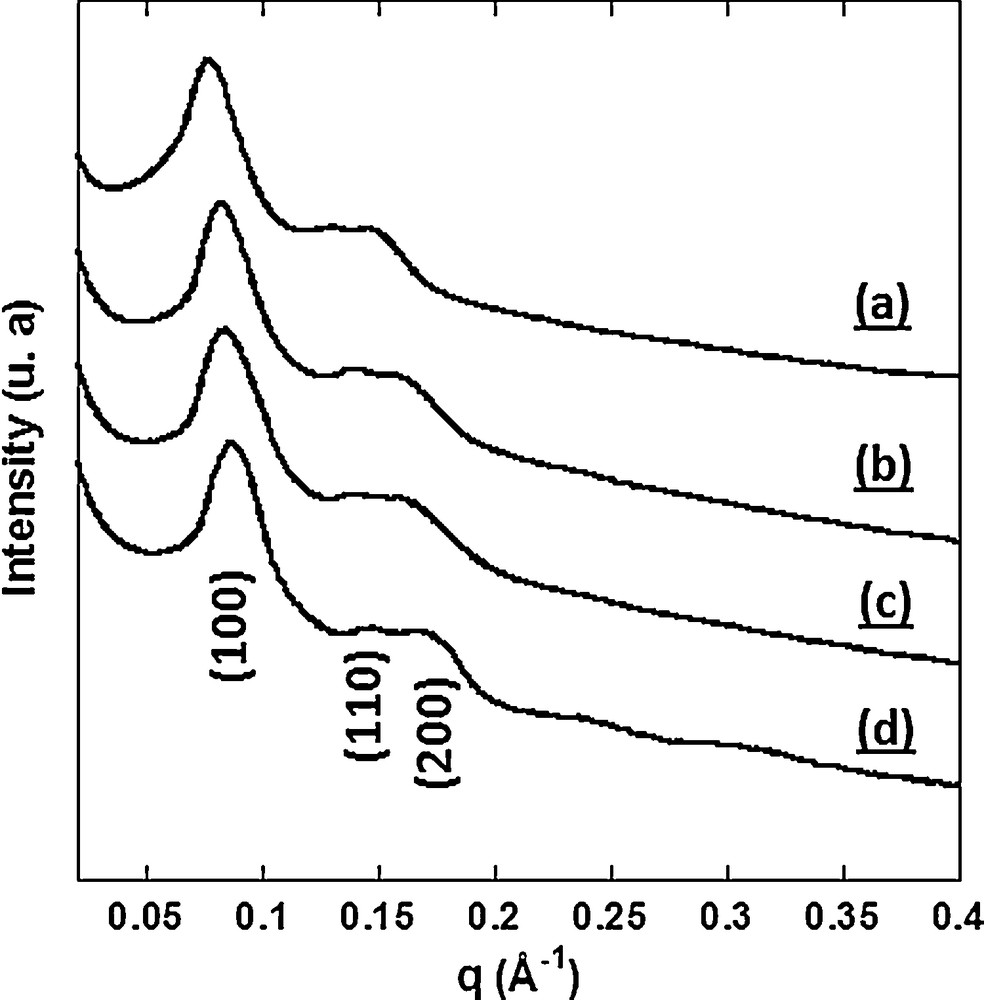
Small-angle X-ray diffraction patterns (a) of Mono CoCo and of Mono CoCo calcined at (b) 600 °C, (c) 700 °C and (d) 800 °C during 2 h.
The diffractograms of Mono CoCo calcined at 600 °C, at 700 °C and 800 °C exhibit three peaks which were indexed as the (100), (110) and (200) reflections of a 2D-hexagonal organization. They are very close to the diffractogram of Mono CoCo, which shows the preservation of the 2D-hexagonal organization after thermal treatment. In addition, a small shift of the peaks towards higher q after thermal treatment indicates a contraction of the lattice due to further silica condensation. A TEM study of Mono CoCo calcined at 800 °C showed the collapse of the ordered porosity in some areas of the sample (Supplementary data, Fig. S4). Such a modification of the silica matrix was not observed for the samples calcined at 600 and 700 °C.
Therefore, the thermal treatment at 700 °C during 2 h was chosen and the product of this treatment is called Mono CoCo–Ox in the following. The same thermal treatment was applied to the powdered CoCo PBA. The calcination product is called CoCo–Ox in the following.
3.2.3 XRD
The diffractograms of CoCo–Ox and Mono CoCo–Ox are shown in Fig. 7. The diffractogram of CoCo–Ox exhibits diffraction peaks, which were indexed within the Fd3m group of the Co3O4 spinel structure. The a cubic lattice parameter was calculated from the position of the (311), (222), (400), (422), (511) and (440) diffraction lines: a = 8.08 ± 0.02 Å.
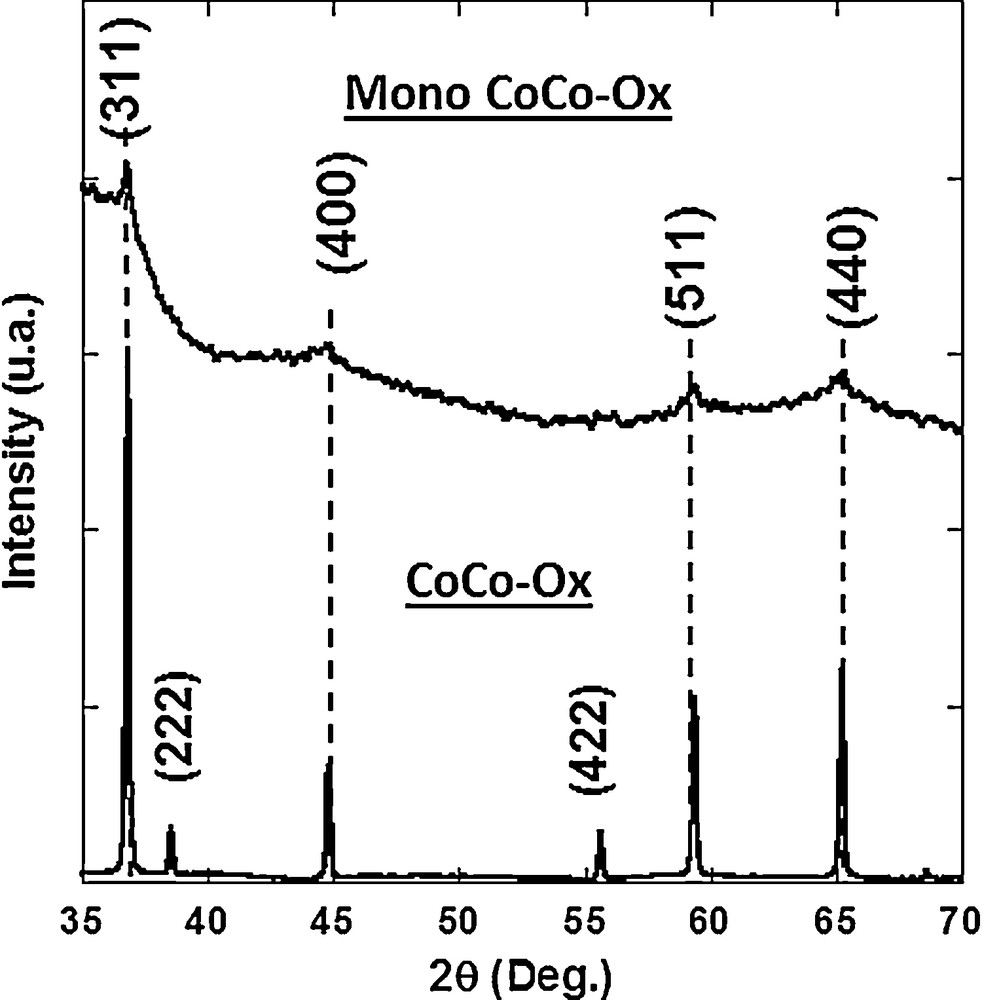
X-ray diffraction patterns of CoCo–Ox and Mono CoCo–Ox.
The diffraction pattern of Mono CoCo–Ox exhibits four broad peaks at the same position than those of CoCo–Ox. They were indexed to the cobalt oxide Co3O4 spinel structure (Fd3m space group). The diffraction peaks are broader and less intense than those of CoCo–Ox. This can be explained by:
- • a smaller amount of Co3O4 in Mono CoCo–Ox than in CoCo–Ox (the nanocomposite contains approximately 4% in weight of Co3O4);
- • a smaller particle size in agreement with the confined transformation of CoCo PBA into the mesopores of the silica matrix.
3.2.4 IR spectroscopy
The IR spectrum of CoCo–Ox is shown in Fig. 8. The absence of bands over the cyanide vibration range indicates the full decomposition of cyanide. The spectrum exhibits two intense absorption bands at 564 cm−1 (ν1) and 670 cm−1 (ν2) assigned to Co–O stretching vibrations in the spinel structure, which confirms the formation of the Co3O4 spinel oxide [39].
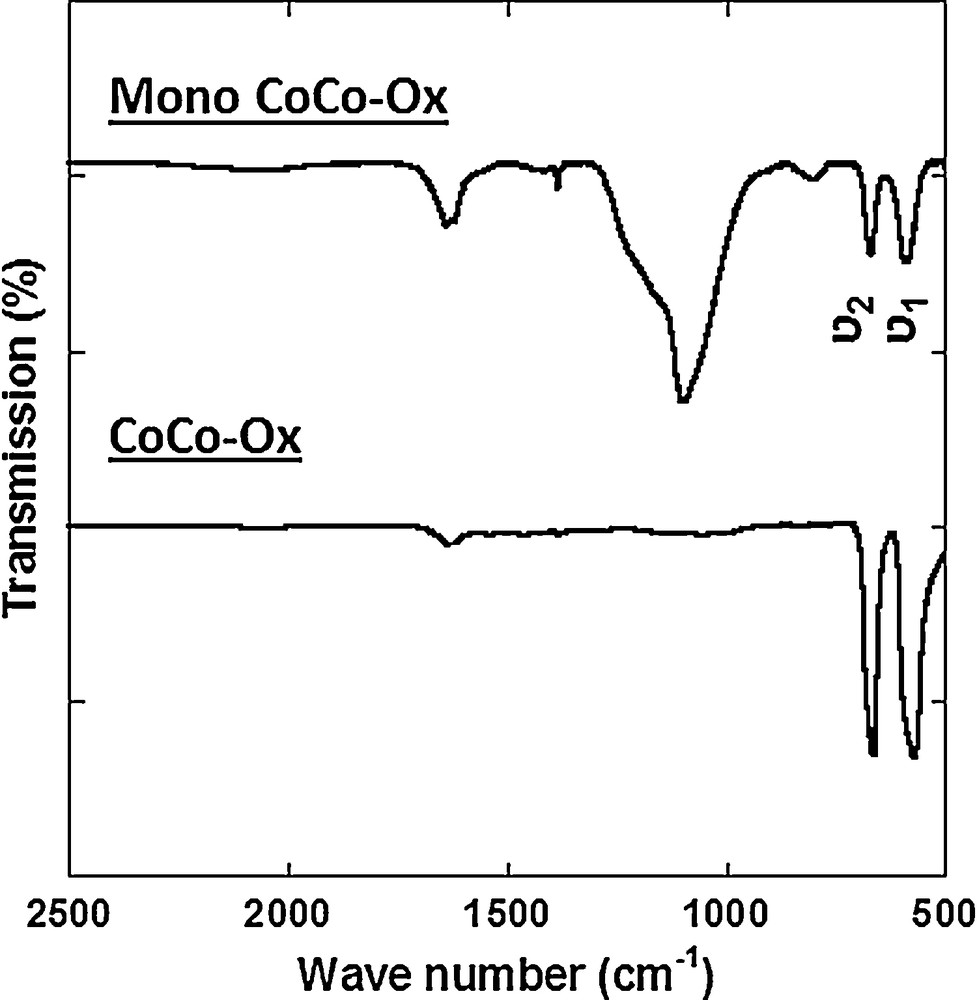
IR spectra of CoCo–Ox and Mono CoCo–Ox after dissolution of the silica matrix.
Fig. 8 shows the IR spectrum of Mono CoCo–Ox after dissolution of the major part of the silica matrix by a KOH concentrated solution. The absence of bands over the cyanide vibration range also indicates the full decomposition of cyanide. The spectrum shows the same absorption bands at 589 cm−1 (ν1) and 670 cm−1 (ν2), which also confirms the formation of the Co3O4 spinel oxide in the nanocomposite [39]. The broader band around 1100 cm−1 is due to residual silica [40].
3.2.5 TEM
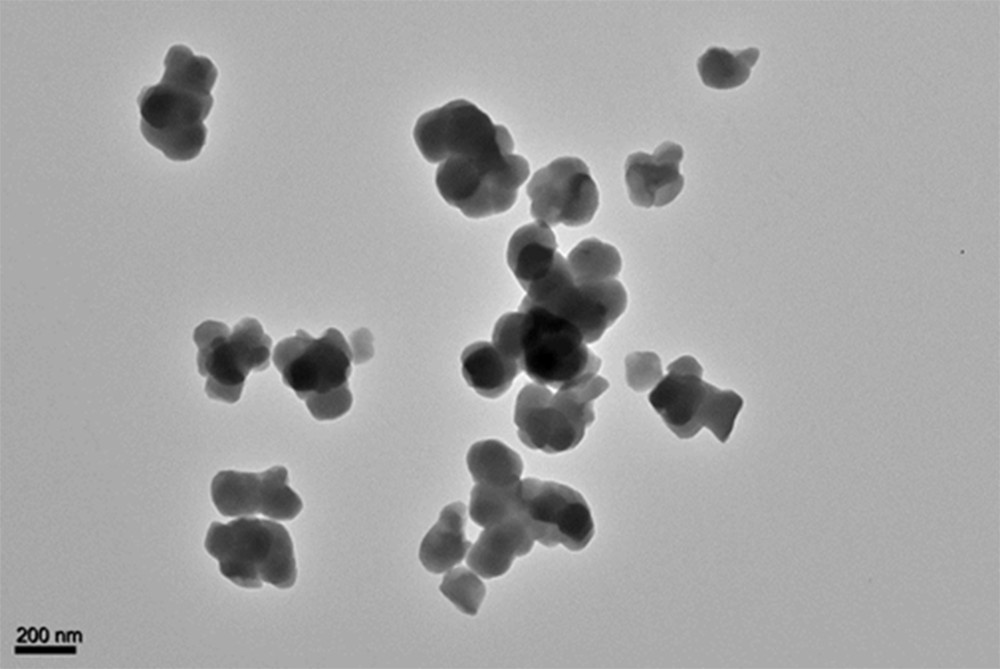
Fig. 9 shows a TEM micrograph of CoCo–Ox. Most particles are in the 300 nm–600 nm size range. They are clearly bigger than the particles constituting the CoCo PBA precursor (CoCo) (Supplementary data, Fig. S2), indicating the occurrence of sintering phenomena during the thermal treatment.

TEM image of CoCo–Ox.
Fig. 10 shows TEM images of Mono CoCo–Ox. Fig. 10(a) and (b) confirms the 2D-hexagonal organization of the porosity. The mesopores are around 5 nm in diameter. No particle was observed outside the silica matrix. Dark spots in the porosity were assigned to oxide nanoparticles. Fig. 10(c) shows a high-resolution micrograph of one Co oxide nanoparticle. This image shows parallel lines corresponding to reticular planes of a single-crystalline particle. The average measured distance between two successive lattice planes (0.245 ± 0.004 nm) of this single-crystal corresponds to an interplanar distance consistent with the (311) d-spacing of Co3O4 spinel oxide. Mono CoCo–Ox can be described as an ordered mesoporous silica monolith containing single-crystalline Co3O4 nanoparticles in its porosity.

TEM micrographs of the 2D-hexagonal Mono CoCo–Ox (a) longitudinal section, (b) transversal section and (c) high resolution TEM micrograph of a Co oxide nanoparticle.
The shape of the particles is mainly spherical and they are isolated from one another but they lie side by side along the mesopores as if they were originating from one PBA nanowire. Particles have an average size of 5 nm in diameter. They fill the entire diameter of the pore, indicating that the size of the particles is limited by the pore size. However, some nanoparticles reach a diameter up to 7 nm. These particles are larger than the diameter of the pores, thus suggesting that the silica walls are pushed away in the course of the Co oxide formation and crystallization.
Thermal treatment of CoCo PBA allowed obtaining well-crystallized Co3O4 spinel structure. Thermal treatment of CoCo PBA confined in the porosity of the silica matrix allowed obtaining single crystals of the same Co3O4 spinel structure. The 2D-hexagonal organization of the silica matrix persists throughout the thermal treatment up to 700 °C. During the thermal treatment, the particles remain well confined in the matrix. Furthermore, silica monoliths act as hard templates allowing the control of the size and the arrangement of the oxide particles.
3.3 Magnetic properties of CoCo–Ox and Mono CoCo–Ox
The molar magnetic susceptibility (χM) of CoCo–Ox is shown in Fig. 11(a). The magnetic behavior of CoCo–Ox is in agreement with that of the anti-ferromagnetic Co3O4 spinel oxide [41,42]. χM exhibits a maximum at TN = 35 K, indicative of an anti-ferromagnetic ordering below TN and a paramagnetic behavior above this temperature. The temperature dependence of χM above 100 K was reproduced by equation (1):
| (1) |
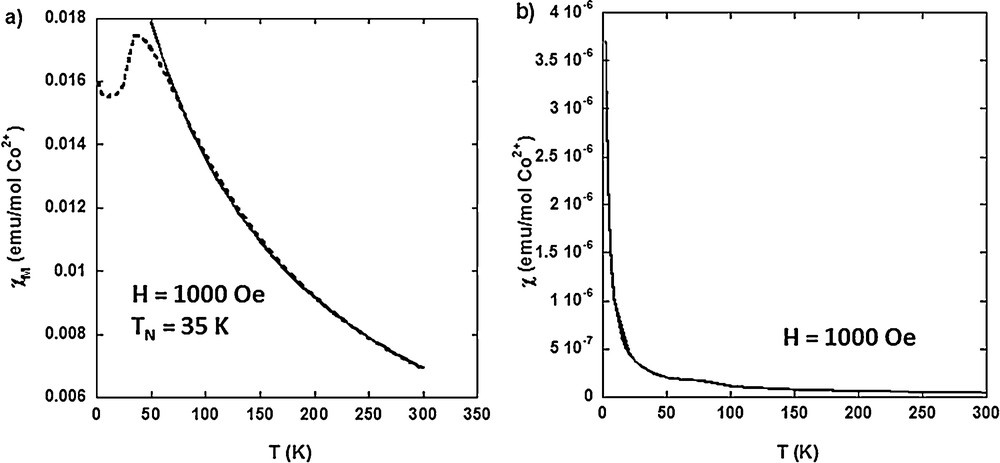
Temperature dependence of (a) the molar magnetic susceptibility (χM) of CoCo–Ox and (b) the magnetic susceptibility (χ) of Mono CoCo–Ox, measured in a 1000-Oe magnetic field.
Fig. 11(b) shows the temperature dependence of the magnetic susceptibility (χ) (reduced magnetization M/H, where H is the applied magnetic field) of Mono CoCo–Ox. The profile of the temperature dependence of the magnetic susceptibility of Mono CoCo–Ox is clearly different from the one of CoCo–Ox. The temperature dependence of χM above 100 K was reproduced by equation (2):
| (2) |
Given the high degree of crystallinity of the particles in both compounds, the different magnetic behaviors can be explained by particle size and/or confinement effects. These magnetic properties are in line with a full confinement of the Co3O4 particles without bigger particles outside. Work is in progress to fully analyze these data.
4 Conclusion
In conclusion, we present here an original process to elaborate oxide nanoparticles using PBA confined in a mesoporous silica matrix as precursor. Starting from monometallic CoCo PBA, we show here that:
- • the PBA-to-oxide transformation is fully confined in the ordered porosity;
- • the oxide nanoparticles are made of single crystals, and their size is determined by the size of the pores.
This control over the crystalline structure and over the particles size at the nanometric scale renders this approach very promising for the elaboration of oxide and mixed oxide nanoparticles with controlled physical properties.
Work is currently in progress to extend this approach to bimetallic PBA in order to elaborate mixed oxide nanoparticles with finely tuned stoichiometry and physical properties.
Acknowledgements
The authors thank D. Durand and E. Rivière for their help in data collection and analysis. Virgile Trannoy thanks Region Ile-de-France C’Nano program for the PhD financial support.
Appendix A Supplementary data
X-ray diffraction pattern of the alkali cation free CoFe Prussian blue analogue after thermal treatment under oxidative atmosphere. TEM micrograph of CoCo PBA (CoCo). TEM micrograph of the Rb2CoCo PBA nanocomposite. TEM micrograph of collapsed area of Mono CoCo after thermal treatment at 800 °C.