

1 Introduction
Ethanol is an environmentally favorable product both as a fuel additive and a hydrogen carrier. Currently, the route of ethanol synthesis from syngas, being encouraged by most of the governments in the world, has not been commercially exploited yet. Rh-based catalysts are the most active ones for the formation of ethanol from CO hydrogenation. Numerous additives, such as alkali metals [1], rare metal oxides [2,3], and transition metal oxides [4–6] have been used in CO hydrogenation for Rh-based catalysts. The support is also an important parameter for the design of catalysts due to its influence on metal dispersion, metal–support interaction [7]. SiO2[8], γ-Al2O3 [9], TiO2 [10], SBA-15 [11], and carbon nanotubes [12] had been used as carriers. However, to date, only the SiO2-supported Rh system has been extensively studied. For example, Goodwin et al. [13] reported that excellent ethanol selectivity was obtained over RhLaFeV/SiO2 catalysts; Mn-, Fe-, Li-promoted Rh/SiO2 catalysts, such as RhMn/SiO2, RhMnli/SiO2 and RhMnFeli/SiO2 have been reported to exhibit good catalytic performances during CO hydrogenation [2]. For TiO2- and Al2O3-supported systems, the commonly reported additives are still Mn and Fe like in RhFe/TiO2 [1], RhFe/γ-Al2O3 [14] and RhMn/γ-Al2O3 [15], suggesting that they are effective promoters. γ-Al2O3 was the most widely used in chemical and petroleum industry as a catalyst carrier [16,17] due to its structure with large pores, its adjustable surface adsorption performance, its good surface acidity and thermal stability. To our knowledge, Mn and Fe co-promoted Rh/γ-Al2O3 have not been reported in CO hydrogenation. In this study, Mn and Fe were chosen as co-promoters and γ-Al2O3 as a carrier. The relations between catalytic structure and activity should be further discussed. In addition, in preliminary experiments, we had screened optimized catalyst compositions of RhMnFe/γ-Al2O3 using an experimental design aiming at maximizing ethanol selectivity with moderate CO conversion. The CO hydrogenation performance of the optimized catalysts (Rh2.5Mn2.5Fe4/γ-Al2O3) and non-promoted or single-element-promoted catalysts (Rh2.5/γ-Al2O3, Rh2.5Mn2.5/γ-Al2O3, Rh2.5Fe4/γ-Al2O3) were investigated in the present study. In an attempt to establish the structure–performance relationships of these catalysts, techniques such as nitrogen sorption measurement, X-ray diffraction (XRD), hydrogen temperature-programmed reduction (H2-TPR) and desorption (H2-TPD), X-ray photoelectron spectroscopy (XPS), CO chemisorption and in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) were used.
2 Experimental
2.1 Catalyst preparation
About 40–60 mesh γ-Al2O3 (∼ 256 m2/g) was used as a carrier. Before being used, it was calcined at 500 °C for 4 h. Rh2.5/γ-Al2O3, Rh2.5Mn2.5/γ-Al2O3, Rh2.5Fe4/γ-Al2O3, and Rh2.5Mn2.5Fe4/γ-Al2O3 catalysts were prepared by a co-impregnation technique as follows. An aqueous solution of Rh(NO3)3 hydrate, Fe(NO3)3 and Mn(NO3)2 was brought into contact with γ-Al2O3, followed by aging at room temperature (RT) for 12 h and then dried at 110 °C overnight before being calcined in the air at 500 °C for 4 h with a heating rate of 2 °C/min. γ-Al2O3 is omitted in the names of the catalysts in figures and tables for simplicity. The numbers after the elements indicate the weight percentage relative to the initial weight of the support material. Also, these numbers will be omitted in the following section.
2.2 Catalyst characterization
The BET surface area, the pore volume and the average pore diameter were determined using N2 adsorption at 77 K after outgassing of the sample under a vacuum of 10−3 mmHg for 4 h at 200 °C in a Micrometric ASAP 2020 automated system. XRD patterns were recorded on a diffractometer operating with Cu Kα radiation at 40 kV. The power diffractograms of the samples were collected from 10 to 80° at a rate of 6°/min. H2-TPR as well as H2-TPD and CO chemisorptions experiments were performed on a Micromeritics Autochem 2920 apparatus with a thermal conductivity detector (TCD). For H2-TPR experiments, the catalyst sample (200 mg) was placed in a quartz U-tube and dried in 50 mL/min Ar at 500 °C for 30 min. After cooling to room temperature, the catalyst was exposed to 50 mL/min of 10% H2/Ar flow. The TPR profile was recorded with a TCD according to H2 consumption, while the sample was reduced from room temperature to 800 °C with a ramp of 10 °C/min. For H2-TPD measurements, about 200 mg of sample was pre-reduced in 50 mL/min of 10% H2/Ar flow at 350 °C for 2 h and was held at the same temperature for another 30 min under an He flow. After cooling to room temperature, H2 was introduced into the catalyst bed until saturation in a pulse mode, and then, the catalyst bed was purged by He for 30 min. Subsequently, the sample was heated up to 800 °C under He flow at a rate of 10 °C/min, while the desorbed products were detected with the TCD detector. In CO chemisorption measurements, the catalyst (50 mg) was reduced as in the procedure for H2-TPD and then cooled to room temperature. After that, CO was passed through the bed in a pulse mode. Then, the physisorbed CO was removed by He for 30 min. The amount of chemisorbed CO and metal dispersion was calculated from the moles of adsorbed CO per total moles of Rh impregnated onto the catalyst support. X-ray photoelectron spectra were recorded over reduced and unreduced catalysts using a VG ESCALAB 250Xi electron spectrometer equipped with a hemispherical analyzer, operating in a constant pass energy mode, and an Al Kα X-ray source operated at 10 mA and 12 kV. The binding energies (BE) were referred to the Al 2p peak. Using this reference, BE values of C 1s peak coming from adventitious carbon appeared at 284.9 ± 0.2 eV. The intensities of the peaks were estimated via integrating each peak after subtracting an S-shaped background and fitting the experimental peak to Lorentzian/Gaussian lines.
In situ DRIFTS was carried out with a spectrometer (Nicolet 6700, MCT detector, Thermo, USA). The catalyst was reduced in H2 for 2 h at 350 °C and then flushed by pure nitrogen for 30 min. The background spectra were collected when the vacuum degree of the in situ cell was better than 10−4 Pa. Then, 5% CO/He was introduced at 30 °C for 30 min. The sample was purged with N2 before the spectra were collected. All spectra were recorded with a resolution of 4 cm−1 and with an accumulation of 64 scans.
2.3 CO hydrogenation
CO hydrogenation of Mn- and Fe-modified Rh/γ-Al2O3 catalysts was carried out in a high-pressure fixed bed under the following reaction conditions: 260 °C, 2.0 MPa, 3600/mL (h·gcat) and H2/CO = 2. Prior to the reaction, 1.0 g of catalyst was reduced at 623 K in flowing H2 for 10 h at 0.1 MPa. The reactor was cooled down to the reaction temperature using an H2 stream, and then gas flow was switched to syngas (H2/CO = 2). The catalyst bed was pressurized to the reaction pressure and the flow rate was controlled using a Brooks 5850 mass flow controller. The tail gas was analyzed online by two sets of GC (Agilent 7890A) in series. The aqueous products were analyzed off-line by GC with an FID detector. The liquid products were collected after 24 h on-stream when the steady state was reached.
3 Results and discussion
3.1 Catalyst characterization
3.1.1 XRD and N2 adsorption
From XRD patterns of the selected samples (not shown), only diffraction peaks corresponding to the support γ-Al2O3 (2θ = 37.39, 45.89 and 67.18°) could be seen, indicating that metal species are highly dispersed on the support or that the size of the crystallite is too small to be examined by XRD. The BET specific surface area, the pore volume and the pore diameter are listed in Table 1. As seen in Table 1, both specific surface area and pore volume decreased with increasing metal loading, while pore diameter were nearly invariable.
Textural properties of the catalysts.
| Catalysts | BET surface area (m2·g−1) | Pore volume (m3·g−1) | Pore diameter (nm) |
| Rh | 219.4 | 0.49 | 7.45 |
| RhMn | 209.5 | 0.46 | 7.42 |
| RhFe | 205.0 | 0.45 | 7.57 |
| RhMnFe | 184.7 | 0.40 | 7.33 |
| γ-Al2O3 | 256.1 | 0.52 | 6.75 |
3.1.2 H2-TPR
H2-TPR results for the above catalysts are shown in Fig. 1. As references, the TPR profiles of Fe/γ-Al2O3 and Mn/γ-Al2O3 were also added in this figure. The unpromoted Rh/γ-Al2O3 presents a broad reduction peak in the range from 80 to 200 °C, with a maximum at about 150 °C. The broad reduction peak observed for Rh/γ-Al2O3 indicates that there should be a sequence of reduction from surface to bulk Rh species. According to H2 consumption, only a portion of rhodium oxide was reduced. Similar observation has been reported by Burch et al. [14], who suggested that some rhodium oxide on the γ-Al2O3 may spread over the support and diffuse into defect sites in the alumina, becoming strongly bound and non-reducible. Fe/γ-Al2O3 presents a broad reduction zone between 236 and 570 °C owing to the reduction of Fe3+ ions to Fe2+ and Fe2+ to Fe0. Mn/γ-Al2O3 shows a reduction peak between 270 and 450 °C as a result of MnO2 → Mn3O4 → MnO [18]. The presence of Mn in Rh/γ-Al2O3 shifted the reduction of Rh species to higher temperatures and in turn facilitated the reduction of Mn species. Bimetallic RhFe/γ-Al2O3 catalyst presents a sharp and strong peak in the temperature range between 100 and 150 °C. The lower temperature and stronger peak did not mean that Fe accelerates Rh reduction. The most likely reason was that some Fe3+ ions were probably reduced into Fe2+ at temperatures under 200 °C and that its peak was overlapped by the reduction peak of Rh3+ [10]. Fe- and Mn-promoted catalysts show a reduction temperature between that of singly Fe- and Mn-promoted catalysts. This phenomenon can be considered as the interaction between Mn, Fe and Rh species.
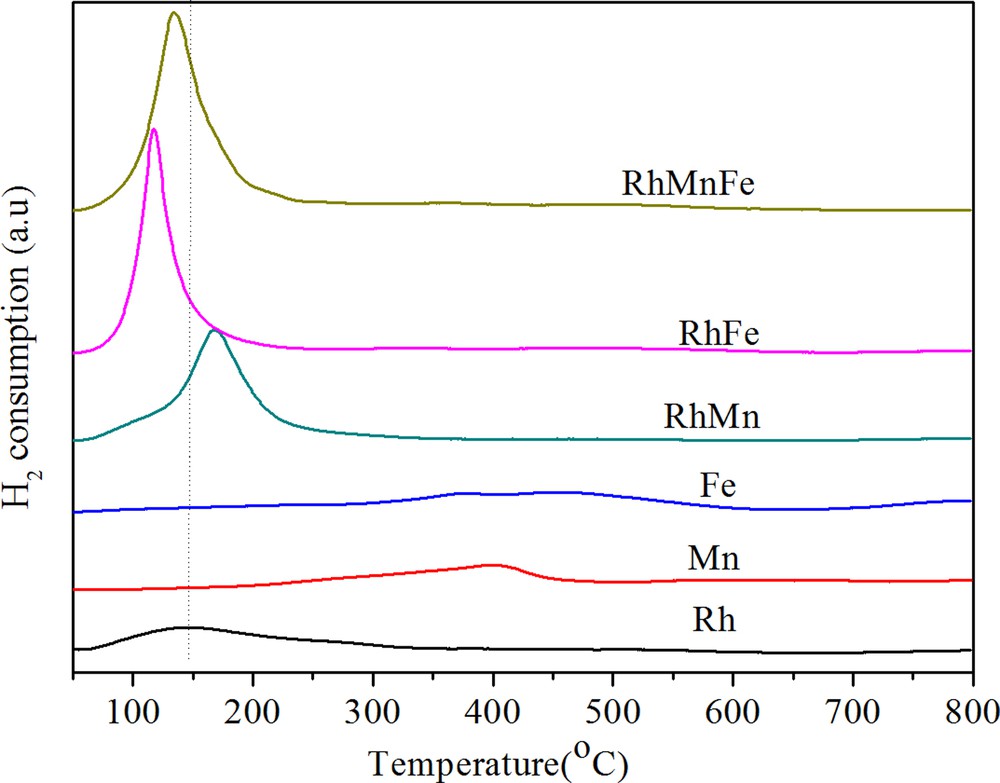
(Color online.) TPR profiles of catalysts.
3.1.3 H2-TPD
H2-TPD can be used to judge the dispersion of Rh catalysts. As can be seen in Fig. 2, the addition of Fe and/or Mn decreased Rh dispersion due to both the low- and high-temperature desorption peak shifted to lower temperature compared to unpromoted Rh catalysts. However, after introduction of Mn into RhFe/γ-Al2O3 catalysts, there existed no apparent decrease in Rh dispersion.
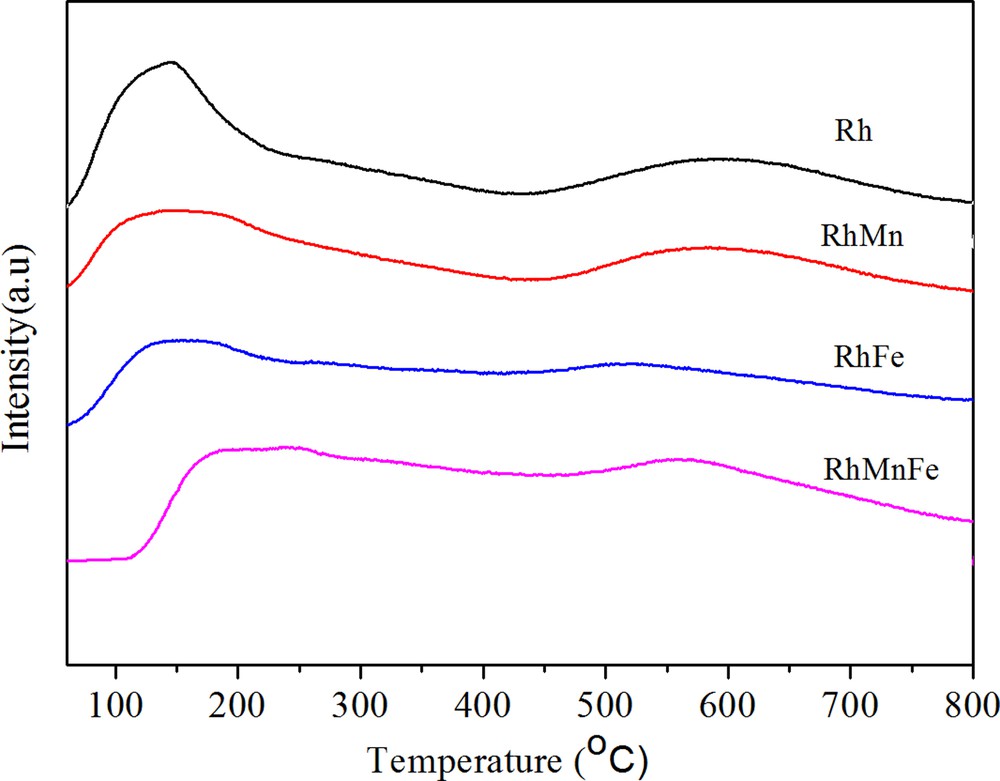
(Color online.) H2-TPD profile of catalysts.
3.1.4 CO chemisorption
Table 2 lists the results obtained from volumetric CO chemisorption for the catalysts. It was found that the addition of Fe resulted in the suppression of CO chemisorption and that Mn just decreased a little, whereas Mn- and Fe-modified Rh/γ-Al2O3 showed significant decrease in CO chemisorption. It was noted that the metal dispersion of the catalyst was not decreased obviously by the addition of Mn to RhFe/γ-Al2O3, which was in line with H2-TPD results, although the dispersion of metal catalysts cannot be fairly accurately estimated by CO or H2 chemisorption [19].
CO chemisorptions on Rh-based catalysts.
| Catalyst | CO-chemisorbed (μmol/g) (total) | Metal dispersion (%) |
| Rh | 193.7 | 79.7 |
| RhMn | 135.3 | 55.7 |
| RhFe | 49.6 | 36.9 |
| RhMnFe | 42.1 | 33.5 |
3.1.5 XPS
XPS studies were carried out to get a deeper insight into the dispersion and the chemical state of Rh on the catalyst surface after calcination and reduction. XPS intensity ratios are listed in Table 3. The results showed that Rh/Al signal ratios for fresh and reduced catalysts decreased in the order of Rh/γ-Al2O3 > RhMn/γ-Al2O3 > RhMnFe/γ-Al2O3 > RhFe/γ-Al2O3, indicating that the addition of Mn to RhFe/γ-Al2O3 did not decrease Rh aggregation. This is in agreement with H2-TPD and CO chemisorption results. In addition, it was observed that a reduced RhMn/γ-Al2O3 catalyst presented higher Mn/Al ratio than calcined ones, while the Fe/Al ratio of RhFe/γ-Al2O3 decreased significantly after reduction. For doubly Mn- and Fe-promoted reduced catalysts, the surface Mn/Al ratio increased and Fe/Al ratio nearly kept unchanged compared to that of the calcined samples, indicating that Mn and Fe interaction resulted in less Mn dispersion and more Fe enrichment on the surface of the catalysts.
Surface composition of catalysts determined by XPS.
| Catalyst | Rh | RhMn | RhFe | RhMnFe | ||||
| Calcined | Reduced | Calcined | Reduced | Calcined | Reduced | Calcined | Reduced | |
| Rh:Al | 0.025 | 0.026 | 0.024 | 0.024 | 0.022 | 0.020 | 0.023 | 0.023 |
| Mn:Al | – | – | 0.025 | 0.051 | – | – | 0.023 | 0.030 |
| Fe:Al | – | – | – | – | 0.081 | 0.043 | 0.066 | 0.067 |
| Rh3+/Rh0 | 0.14 | 0.32 | 0.88 | 0.72 |
Fig. 3(top) displays the Rh3d spectra after calcination. The binding energies were calibrated relatively to the C 1s peak from carbon contamination of the samples at 284.8 eV. In all cases, the Rh3d5/2 level was located at 309.1–309.9 eV, and ascribed to Rh3+. The BE of RhMnFe/γ-Al2O3 was 309.2 eV, lower than that of 3d5/2 energy level of Rh/γ-Al2O3 (309.7 eV) or RhMn/γ-Al2O3 (309.5 eV), but higher than that of RhFe/γ-Al2O3 (309.1 eV), which may have been caused by an electronic modification of the rhodium by Rh–M or Rh–M–M interaction (M = Mn or Fe). The chemical changes in the rhodium species after reduction in H2 at 623 K were also investigated by XPS. As shown in Fig. 3(bottom), the Rh3d5/2 peak of reduced samples could be resolved into two components with binding energies of 307.3–307.6 eV and 308.8–309.3 eV. The former corresponded to the Rh0 species and the latter could be attributed to Rh3+ [20]. These results suggested that Rh0 co-existed with oxidized Rh species on the surfaces of all the reduced catalysts. The ratios of energy region area of Rh3+ and Rh0 (being overshadowed) are listed in Table 3, suggesting that Rh3+ on the surface of a single Fe-promoted Rh catalyst was more difficult to get reduced than that on double Mn- and Fe-promoted catalysts.
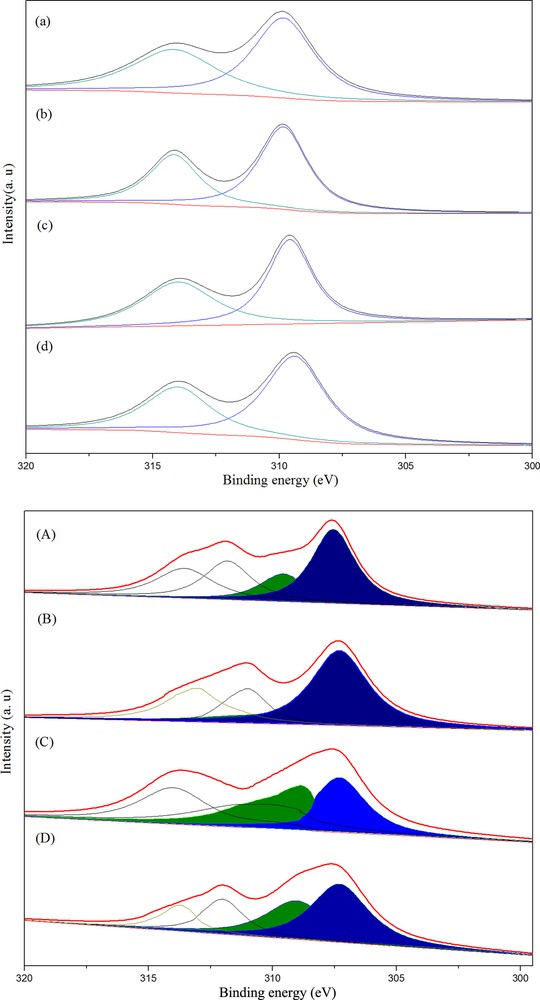
(Color online.) Deconvoluted Rh 3d (XPS) spectra of calcined (top) and reduced (bottom) catalysts ((a), (A), Rh2.5; (b), (B), Rh2.5Mn2.5; (c), (C), Rh2.5Fe4; (d), (D), Rh2.5Mn2.5Fe4).
3.1.6 DRIFTS
Fig. 4 gives the DRIFTS spectra obtained after CO adsorption at 30 °C on various Rh/γ-Al2O3 catalysts. All the catalysts analyzed exhibited three main bands in the metal carbonyls region, namely, the band around 2050 cm−1, the doublet at 2086 and 2012 cm−1 and a broad band centered at 1860 cm−1, with different intensities. The 2050 cm−1 band was attributed to the linear adsorbed CO (CO (l)) on Rh0. The negligible change of the linear CO frequency indicated that the electronic structure of Rh was not altered by the promoter. The doublet can be ascribed to the symmetric and asymmetric carbonyl stretching of the gem-dicarbonyls Rh+ (CO)2(CO(gdc)) on Rh+. Ichikawa et al. proposed that Rh0 is active for CO dissociation, Rh+ is favorable for CO insertion to form intermediates of C2 oxygenates [21]. Many subsequent authors supported this mechanism and tried to correlate activity data with Rh+ or the ratio of Rh+/Rh0. However, some authors argued that only Rh0 was active for oxygenate formation [22]. Our results from XPS indicated that both oxidized Rhδ+ and metallic Rh0 species were present on the catalyst surface after reduction by H2 at 350 °C. Furthermore, according to H2 consumption of TPR profile, Rh3+ ions were not able to be fully reduced. Thus, it can be demonstrably concluded that Rhδ+ existed on the surface of the Rh catalysts after reduction. The board band (1750–1930 cm−1) centered at 1860 cm−1 was assigned to bridge-bonded CO (CO (b)). It is noteworthy that the range of the CO (b) band on these catalysts was far more broad (1740–1930 cm−1) than that (1840–1860 cm−1) on SiO2-supported Rh catalysts. This difference may be due to a different intensity of the Rh–CO band as a result of the promoter–support effect. In our study, it was found that the formation of CH4 was mainly related to the intensity of CO (b) due to the CH4 selectivity, which decreased with decreasing the intensity of CO (b). CO (b) was also formed on Rh0 sites and CH4 could be more easily formed on bridge CO than linear CO [22]. It was observed that the addition of Fe significantly suppressed CO adsorption on Rh/γ-Al2O3 and that the addition of Mn slightly suppressed CO adsorption, while Mn- and Fe-promoted catalysts exhibited the weakest CO adsorptions.
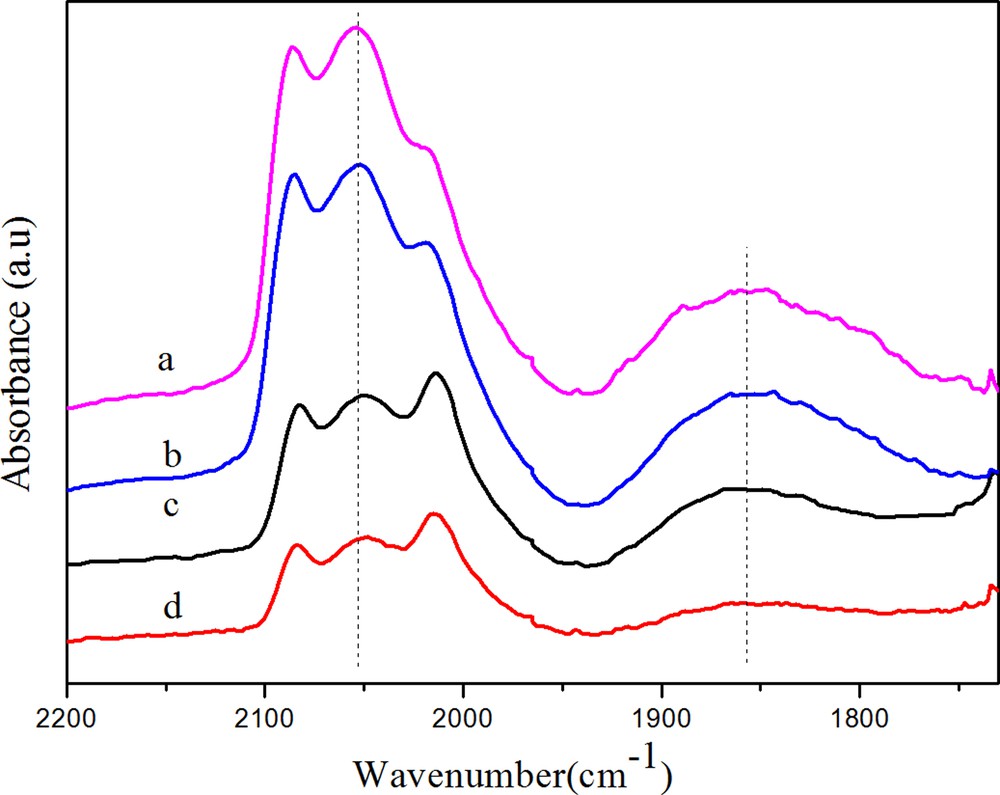
(Color online.) DRIFTS spectra of adsorbed CO at 30 °C on catalysts (a, Rh; b, RhMn; c, RhFe; d, RhMnFe).
Fig. 5 shows the IR spectra of CO adsorbed on the catalysts as a function of temperature in the presence of hydrogen. It can be seen that for unpromoted Rh catalysts, the gem-dicarbonyl CO was gradually weakened with the increase of temperature and disappeared at 230 °C in the H2 stream. The disappearance of CO (gem) may be ascribed to its low thermal stability or to the reduction of Rh+(CO)2 to form CO2 and Rh0x(CO) [23]. However, CO (gem) still existed at 260 °C for RhMn/γ-Al2O3 and could be observed at 230 °C for RhFe/γ-Al2O3 or RhMnFe/γ-Al2O3. This indicated that both Mn and Fe could stabilize CO (gem) species compared to unpromoted Rh/γ-Al2O3. For all catalysts, CO (l) was dominating at the reaction temperature (260 °C) and shifted to lower temperature, which also was observed on some reported catalysts [24]. This shift may be due to a decreasing coverage [1]. In addition, CO (l) got weakened on Rh/γ-Al2O3 and RhMn/γ-Al2O3 compared with RhFe/γ-Al2O3 or RhMnFe/γ-Al2O3. This suggested that Fe could also stabilize the adsorbed CO (l). The bridge-bonded CO on Rh/γ-Al2O3 was observed at 260 °C, while it almost disappeared at 100 °C for Mn and Fe-promoted catalysts. This phenomenon may be attributed to the addition of promoters weakening the bridge bond intensity between CO and Rh (Figs. 6–8).
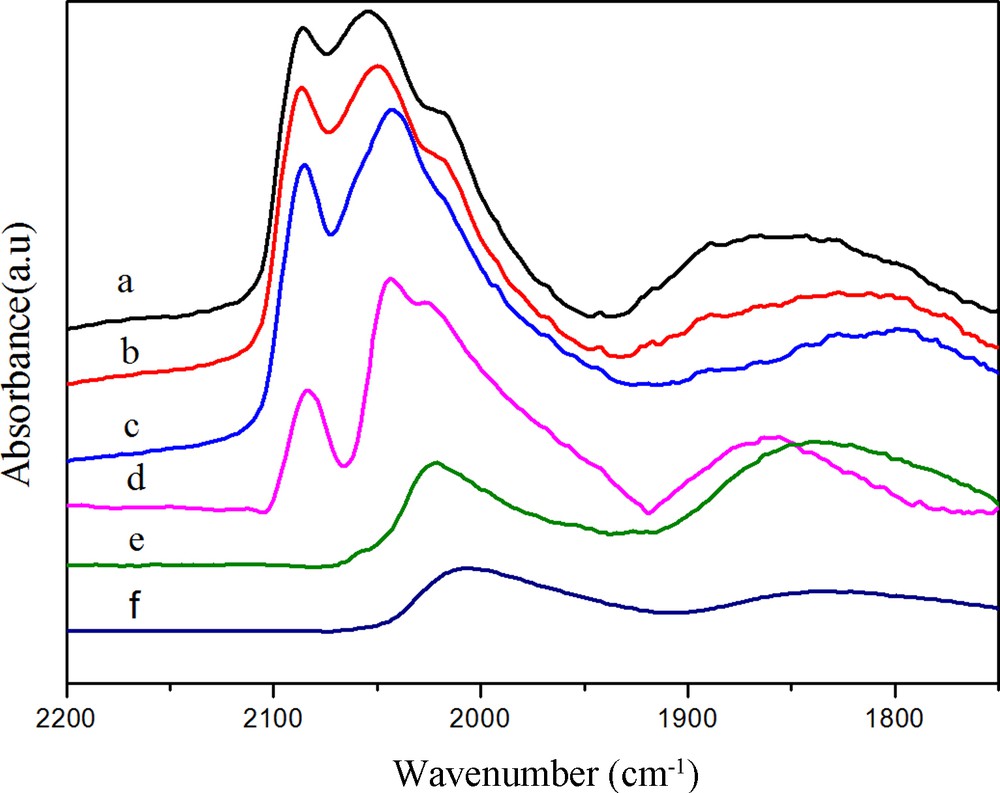
(Color online.) DRIFTS spectra of adsorbed CO at 30 °C on Rh (a) after purging in N2 for 30 min, (b) after heating in H2 at 50 °C, (c) after heating in H2 at 100 °C, (d) after heating in H2 at 180 °C, (e) after heating in H2 at 230 °C, (f) after heating in H2 at 260 °C.
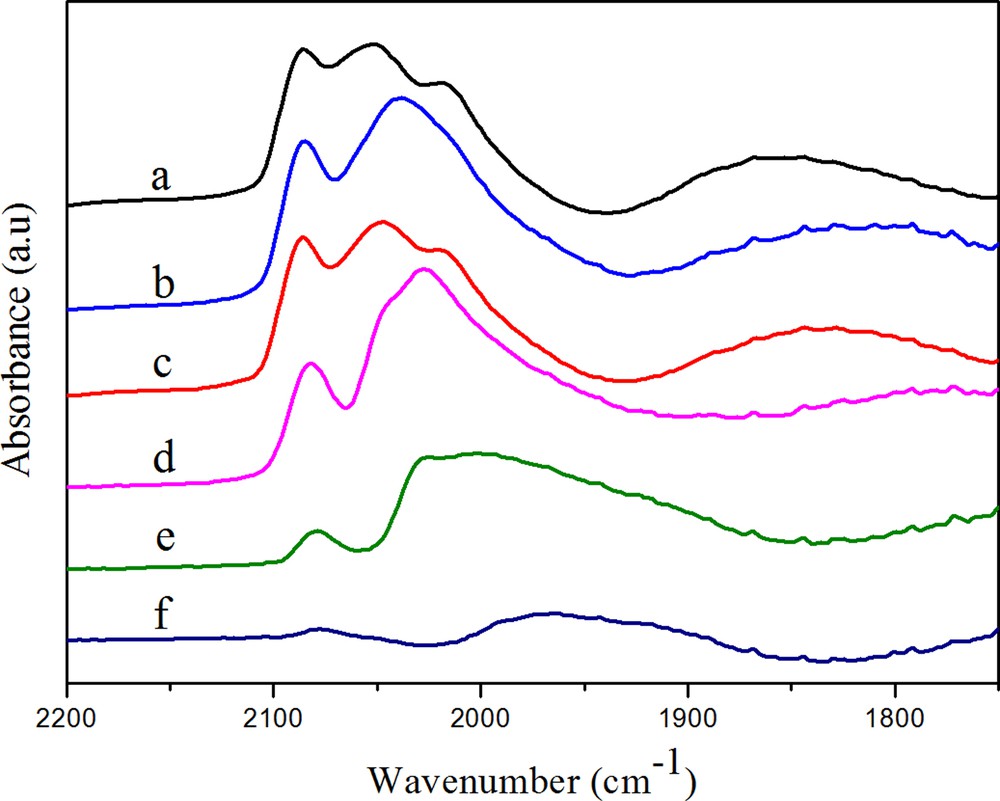
(Color online.) DRIFTS spectra of adsorbed CO on RhMn (a) after purging in N2 for 30 min, (b) after heating in H2 at 50 °C, (c) after heating in H2 at 100 °C, (d) after heating in H2 at 180 °C, (e) after heating in H2 at 230 °C, (f) after heating in H2 at 260 °C.
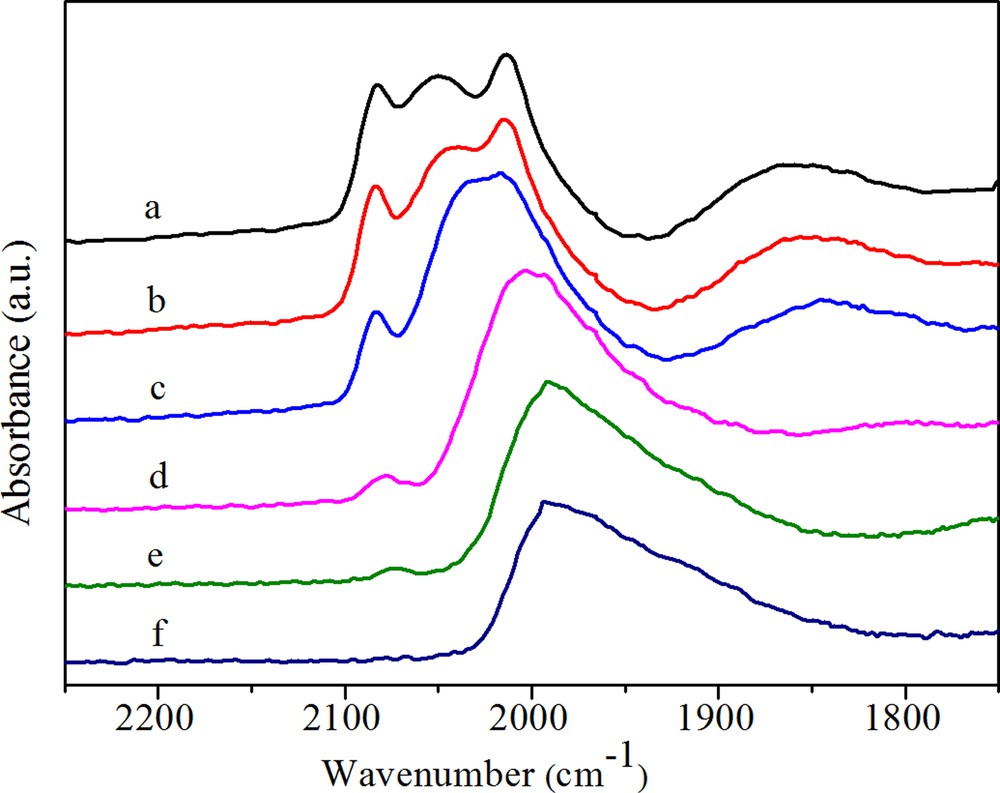
(Color online.) DRIFTS spectra of adsorbed CO on RhFe (a) after purging in N2 for 30 min, (b) after heating in H2 at 50 °C, (c) after heating in H2 at 100 °C, (d) after heating in H2 at 180 °C, (e) after heating in H2 at 230 °C, (f) after heating in H2 at 260 °C.
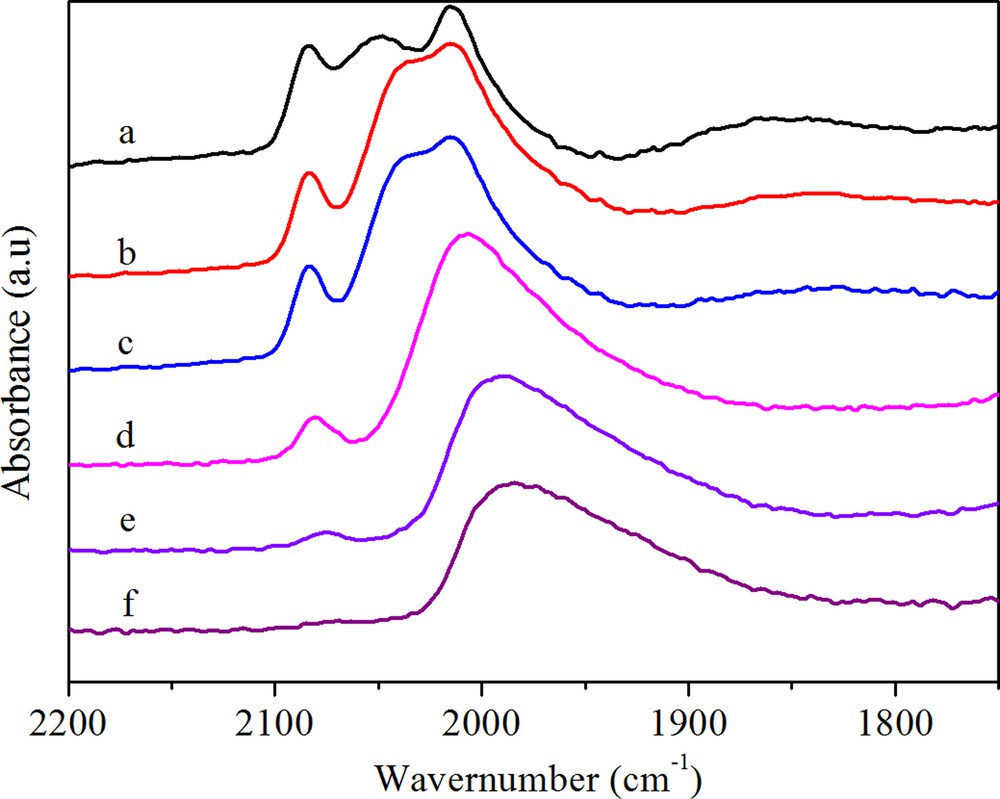
(Color online.) DRIFTS spectra of adsorbed CO on RhMnFe (a) after purging in N2 for 30 min, (b) after heating in H2 at 50 °C, (c) after heating in H2 at 100 °C, (d) after heating in H2 at 180 °C, (e) after heating in H2 at 230 °C, (f) after heating in H2 at 260 °C.
3.2 CO hydrogenation results
Fig. 9 shows CO conversion as a function of the time of stream (TOS) for various catalysts. A deactivation behavior was observed at the initial stages for all the catalysts. Fe-promoted Rh/γ-Al2O3 catalysts required longer time to reach a steady state. Double Mn- and Fe-promoted catalysts got steady within a shorter time than with RhFe/γ-Al2O3, indicating that the interaction between Mn and Fe may have an effect on the stabilization of active species to some degree.

(Color online.) TOS of the catalysts.
Table 4 lists the results of CO hydrogenation over various catalysts. It can be seen that Fe-promoted catalysts exhibited higher CO conversion and a more moderate selectivity than unpromoted ones. On the other hand, single Mn-promoted Rh catalysts showed a lesser increase than Rh/γ-Al2O3. It was reported by Ojeda et al. [15] that the addition of Mn to Rh/γ-Al2O3 improved ethanol selectivity. However, our experimental results showed that RhMn/γ-Al2O3 did not result in an increase in ethanol selectivity. This may be attributed to different reduction conditions, precursor, preparation process and textual properties of γ-Al2O3. RhMnFe/γ-Al2O3 resulted in higher CO conversion, higher ethanol selectivity compared to single-metal or unpromoted Rh catalysts. It was evident that the RhMnFe/γ-Al2O3 produced lower methane and methanol compared to the RhFe/γ-Al2O3. This manifested that the incorporation of Mn to RhFe/γ-Al2O3 decreased the hydrogenation ability of adsorbed non-dissociative CO and CHx and thus provided more chance for CO insertion. It has been reported that (Rhx0–Rhy+)–O–M is the active site for the formation of ethanol [25]. The promoting effect of Fe was reported to be related to the oxidation state of Fe [26]. The authors suggested that Fe0 could increase catalytic activity, while Fe3+ (Fe2+) could increase selectivity. Our DRIFTS experiments results showed that a larger number Rh0 species remained at reaction temperature for RhFe/γ-Al2O3 or RhMnFe/γ-Al2O3. Furthermore, according to H2-TPR and XPS results, the addition of Mn to RhFe/γ-Al2O3 may play a role in the inhibition of Fe3+ reduction. Therefore, one of the reasons for higher ethanol selectivity over the double promoted catalyst may be due to that this catalyst owned more (Rhx0–Rhy+)–O–Fe3+(Fe2+) active sites. The synergistic promoting effect of the combined manganese and iron addition through intimate contact with Rh weakened the CO bond and activated the adsorbed CO species, which may be another reason for its good performance over RhMnFe/γ-Al2O3.
Results of CO hydrogenation over various catalystsa.
| Catalysts | XCO (%) | Product selectivity (%) | |||||
| CH4 | C2+HC | MeOH | EtOH | CO2 | Otherb | ||
| Rh | 11.4 | 40.1 | 22.0 | 8.9 | 12.0 | 9.9 | 6.1 |
| RhFe | 18.9 | 25.4 | 12.6 | 22.9 | 18.7 | 17.0 | 3.3 |
| RhMn | 12.5 | 41.3 | 15.7 | 6.9 | 12.3 | 14.8 | 8.9 |
| RhMnFe | 22.8 | 19.6 | 10.2 | 16.8 | 27.8 | 15.4 | 10.2 |
a Catalyst: 1 g; reaction conditions: P = 2 MPa, H2/CO = 2, space velocity = 3600 mL/(h·gcat).
b Oxygenates with two or more carbons except ethanol (acetaldehyde, acetone, n-propanol, i-propanol, n-butanol, i-butanol and n-pentanol).
4 Conclusions
The direct synthesis of ethanol from CO hydrogenation over Mn and/or Fe-promoted Rh/γ-Al2O3 was explored. The results showed that Fe addition improved CO conversion and increased ethanol selectivity. The results showed that both CO conversion and ethanol selectivity were improved distinctly by the addition of Fe. A significantly enhanced ethanol selectivity and a slightly increased CO conversion were presented on the Mn modified RhFe/γ-Al2O3. Various characterizations indicated that Fe can produce more obvious inhibiting effects on Rh dispersion, reduction and CO adsorption than Mn. DRIFTS investigations indicated that higher CO conversion and ethanol selectivity over RhMnFe/γ-Al2O3 may be attributed to more (Rhx0–Rhy+)–O–Fe3+(Fe2+) active sites.
Acknowledgements
This work is financially supported by the State Science & Technology Support Program of China (No. 2007BAA08B04).