

1 Introduction
The Knoevenagel condensation, one of the most important reaction for the formation of CC bond between carbonyl functionality and active methylene compound, has been widely used to prepare versatile fine chemical intermediates, natural and therapeutic drugs, polymers, cosmetics and perfumes [1]. Traditionally, it is catalyzed by some acids, organic bases as well as their salts, including dimethylamino pyridine, piperidine, guanidine, ethylenediamine [2]. However, the tedious recovery of these catalysts reaction solutions imposed environmental barrier upon their further application in industrial scale. Recently, some heterogeneous Lewis acids [3], sulfate-ion promoted Zirconia [4], layered double hydroxides [5], amine functionalized polyacrylonitrile fiber [6], Ni-SiO2 [7], Si-MCM-41 supported basic materials [8] and task-specific ionic liquids [9] have been explored as catalysts for the Knoevenagel condensation and more or less success has been achieved. Unfortunately, the use of hazardous and carcinogenic solvents and relatively lower catalytic activities with regard to heterogeneous catalysts in the above methodologies are incompatible with “green chemistry”. In accordance with the principles of “green chemistry”, the requirement of the design of catalysts for Knoevenagel condensation with readily separation, highly catalytic activity, low cost is still highly needed.
Magnetic nanoparticles (MNPs) supported catalysts, exhibiting their intrinsically high surface area, readily dispersion in reaction solution and paramagnetic properties, mimic their homogeneous counterparts and, at the same time, enable the trouble-free separation of the catalyst from the reaction mixture simply using by an external magnet [10]. Unmodified MNPs are reactive in acid solution to lose their magnetic properties and tend to aggregate into large clusters because of their anisotropic attraction. In order to overcome these problems, silica was used to coat the MNPs to form a core-shell structure (Fe3O4), which also provided numerous surface Si-OH groups for further versatile modifications [11]. Thus, many silica coated MNPs (SiO2@Fe3O4) supported catalysts have been developed and successfully explored in a range of organic reactions, demonstrating the excellent catalytic activities in Suzuki, Heck and Sonogashira coupling reactions [12], enantioselective asymmetric hydrogenation of aromatic ketones [13], one-pot synthesis of benzoxanthenes [14], aerobic oxidation of alcohols [15], reduction of α,β-epoxy ketones [16].
Ultrasonic irradiation has been frequently utilized in various organic transformations [17]. Cativation process that include the formation, growth, collapse of millions of bubbles within very short time, which induces very high local temperature and pressure, then strengthen the mass transfer between the vapor bubble and ambient liquid [18]. The distinctive behavior of ultrasonic irradiation renders ultrasonic assisted reaction have some advantages, such as short reaction time, improved selectivity, high yield and clean reaction. Driven by the unique properties of magnetic nanoparticles, their successful utilization in organic transformations and significant promotion of ultrasonic technique for organic reactions, herein we report a simple silica coated magnetic nanoparticle supported amine (SiO2@MNP-A) catalyzed, ultrasound promoted, aqueous Knoevenagel condensation of aromatic aldehydes and α-aromatic (heteroaromatic or polyaromatic)-substituted methylene compounds (Scheme 1), in which the active carbon exhibit relatively weak nucleophilicity because of the steric hindrance of aryl group [19].
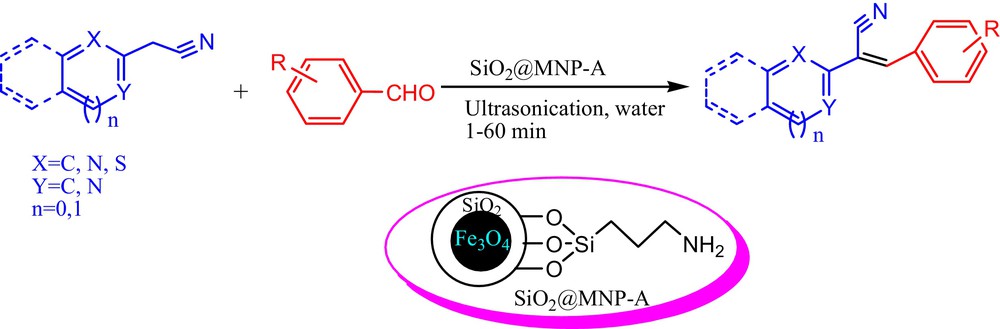
(Color online.) Knoevenagel condensation of aromatic aldehydes and α-aromatic substituted methylene compounds catalyzed by amine functionalized silica coated Fe3O4 nanoparticles (SiO2@MNP-A).
2 Results and discussion
2.1 Preparation and characterization of the catalyst (SiO2@MNP-A)
The magnetic nanoparticle supported amine catalyst (SiO2@MNP-A) was prepared via the procedure depicted on Scheme 2. The magnetic nanoparticles (MNPs) were prepared by the chemical coprecipitation methods [20]. In order to avoid the aggregation problem of naked nano-Fe3O4, an outer silica shell, also providing the suitable sites (Si-OH) for surface functionalization with amine, was coated on the MNP through the ultrasound promoted reaction of highly dispersing MNP with tetraethoxysilane (TEOS) under basic conditions [21].

Synthesis of the magnetic nanoparticle supported amine catalyst.
To guarantee the successful immobilization of amine on the silica coated Fe3O4 nanoparticles, FT-IR was used to investigate the blank MNP (SiO2@MNP) and the catalyst SiO2@MNP-A. On Fig. 1, the bands at 579 cm−1 were assigned to vibration of the FeO bonds, which show the existence of the Fe3O4 ingredients in two samples. Two peaks at 1099 cm−1, 964 cm−1 can be attributed the SiOSi stretching vibration of silica shell. As shown on Fig. 1, the two examples both have broad peak at 3447 cm−1, which was the characteristic of the SiOH stretching vibration. In the curve of SiO2@MNP-A, two obvious bands 1211 cm−1, 1153 cm−1 were verified as CN bond vibration. At the same time, there are two weak peaks at 2949 cm−1 and 2872 cm−1, which were assigned to the CH stretching vibration of alky chain of amine. No absorbance at these wave numbers in the curve of SiO2@MNP was observed, which demonstrating the successful immobilization of amine on the surface of the silica coated Fe3O4 particles.
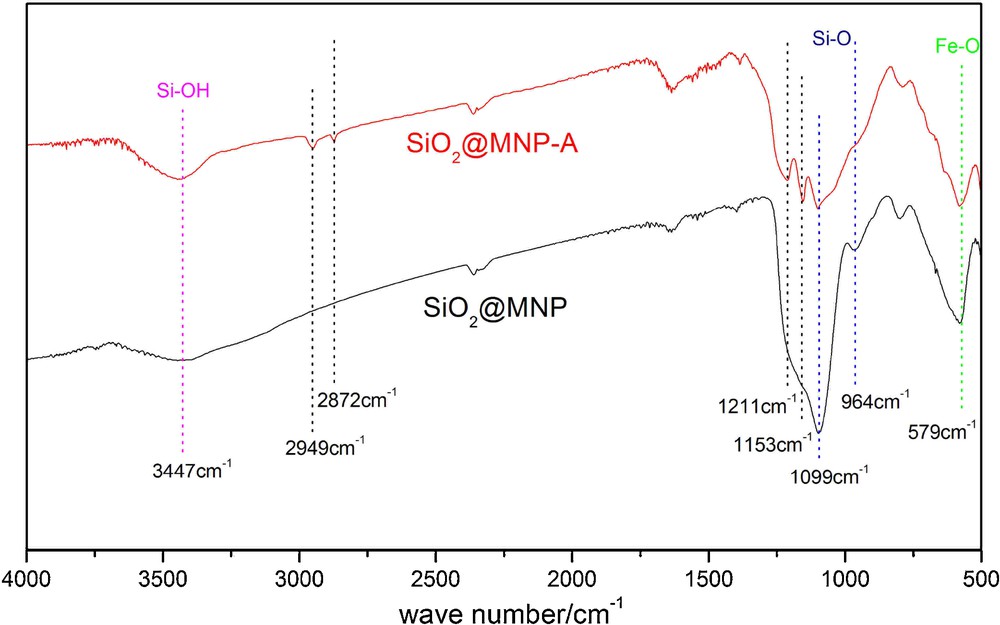
(Color online.) FT-IR spectra of SiO2@MNP and amine functionalized silica coated Fe3O4 nanoparticles (SiO2@MNP-A).
The crystalline structure of SiO2@MNP was characterized by X-ray diffraction (XRD). The diffraction pattern gives characteristic peaks and relative intensity matched well with the standard Fe3O4 nanoparticles (JCPDS file No. 19-0629, Fig. 2). The broad peak from 2θ = 20° to 30° shows the typical properties of amorphous silica phase [22], verifying the successful coat of SiO2 on the Fe3O4 particles. Moreover, transmission electron microscope (TEM) analysis displays that the particle size of the catalyst SiO2@MNP-A is similar to that of blank carrier SiO2@MNP (Fig. 3), with the average size range of 20–30 nm for dark Fe3O4 core which is surrounded by a grey silica shell about 4–6 nm thick. The loading amount of amine was determined to be 0.75 mmol g−1 by elemental analysis.

(Color online.) X-ray diffraction pattern of SiO2@MNP.

Transmission electron microscope images of SiO2@MNP (a) and amine functionalized silica coated Fe3O4 nanoparticles (SiO2@MNP-A) (b).
The magnetic behavior of the neat SiO2@MNP and the catalyst SiO2@MNP-A was investigated using Magnetic Properties Measurement System-5 (MPMS-5, Quantum Design) magnetometer. As shown on Fig. 4, the field-dependent magnetization curves at ambient temperature reveal that SiO2@MNP and SiO2@MNP-A are both superparamagnetic with saturation magnetizations of 15.2 emu g−1 and 6.1 emu g−1, respectively. No phenomenon of hysteresis is detected from the two curves, further proving their superparamagnetic characteristics.

(Color online.) Magnetic curves of SiO2@MNP and amine functionalized silica coated Fe3O4 nanoparticles (SiO2@MNP-A).
2.2 SiO2@MNP-A catalyzed Knoevenagel condensation of aromatic aldehydes and α-aryl -substituted methylene compounds in water under ultrasonic irradiation
In an initial screening of solvents under ultrasonic irradiation (250 W), while dichloromethane (CH2Cl2), toluene and chloroform (CHCl3) showed not to be effective for the reaction of benzaldehyde and benzothiazole-2-acetonitrile, methane (CH3OH), ethanol (C2H5OH) and water provided the final product with promotional value of yield (Table 1, entries 1–6). From the viewpoints of environmental benign friendliness and cost efficiency, water was the optimal choice of solvent for further examinations. The influence of ultrasound power was also investigated. As shown in Table 1, the best yield was obtained by ultrasonic irradiation for 10 min at room temperature and 200 W. Significantly, decreased yields were detected when the power was decreased from 200 W to 50 W (Table 1, entries 8–10). On the contrary, the reaction times and yields did not change from 200 W to 300 W. Thus, 200 W of ultrasonic irradiation was enough to push the reaction forward (Table 1, entries, 3, 10, 11). Without ultrasonic irradiation, only 35% yield of desired product was obtained even if the reaction time was prolonged to 2 h (Table 1, entry 7).
The effect of solvent (20 mL) on the reaction of benzaldehyde (10 mmol) and benzothiazole-2-acetonitrile (10 mmol) in the presence of catalyst SiO2@MNP-A at room temperature under ultrasonic irradiation.
| Entry | Solvent | Power (W) | Time (min) | Yielda |
| 1 | Methanol | 250 | 10 | 90 |
| 2 | Ethanol | 250 | 20 | 86 |
| 3 | Water | 250 | 10 | 94 |
| 4 | CH2Cl2 | 250 | 20 | 72 |
| 5 | Toluene | 250 | 18 | 63 |
| 6 | CHCl3 | 250 | 25 | 74 |
| 7 | Water | 0 | 2 h | 35 |
| 8 | Water | 50 | 1 h | 58 |
| 9 | Water | 150 | 30 | 68 |
| 10 | Water | 200 | 10 | 94 |
| 11 | Water | 300 | 10 | 93 |
a Isolated yield based on benzaldehyde.
The same model reaction of benzaldehyde and benzothiazole-2-acetonitrile under ultrasonic irradiation was exploited to optimize the catalysts as well as their loading. From the results provided in Table 2, it is obvious that the catalyst SiO2@MNP-A worked well to give the desired product within 40 min in 93% yield at room temperature under ultrasonic irradiation (Table 2, entry 7). For comparison, the support SiO2@MNP and silica supported amine (SiO2@A) [23] were investigated as catalysts for the model reaction and the corresponding product was obtained in 15% and 63%, respectively (Table 2, entries 1–2). In addition, organic base propylamine gave the product yield of 82%, which is lower than that catalyzed by SiO2@MNP-A (Table 2, entries 3 and 7). These results confirmed that SiO2@MNP-A performed more effectively as catalyst than the homogeneous organic base, probably due to the absorption of reaction substrates on the surface of the nanoparticles, which lead to the increase in the local concentration of reactants around the active site of the silica coated Fe3O4 catalyst. To find an optimal loading amount of catalyst loading, the model reaction was carried out in the presence of the various amount of SiO2@MNP-A (Table 2, entries 5–8). The rational amount of the catalyst was 2.0 mol %.
Effect of catalyst and loading on the Knoevenagel condensation of benzaldehyde (10 mmol) and benzothiazole-2-acetonitrile (10 mmol) in water (20 mL) under ultrasonic irradiation (200 W) at room temperature.
| Entry | Catalyst (mol %) | Time (min) | Yield (%)a |
| 1 | SiO2@MNP (0.4 g) | 20 | 15 |
| 2b | SiO2@A (3.0) | 20 | 63 |
| 3 | Propylamine | 20 | 82 |
| 4 | – | 30 | Trace |
| 5 | SiO2@MNP-A (1.0) | 10 | 58 |
| 6 | SiO2@MNP-A (1.5) | 10 | 81 |
| 7 | SiO2@MNP-A (2.0) | 5 | 93 |
| 8 | SiO2@MNP-A (2.5) | 10 | 94 |
a Isolated yield based on benzaldehyde.
b SiO2@A: 3-amino-propylated silica gel [21] and the loading amount was 65 mmol g−1.
To evaluate the scope and limitations, as well as the role of ultrasonic irradiation in this methodology, we extended our studies to the reaction of benzothiazole-2-acetonitrile and a variety of structurally diverse aldehydes in the presence of 2.0 mol % SiO2@MNP-A under various conditions, including room temperature stirring (rt), conventional heating (60 °C) and ultrasonic irradiation (US). The results are summarized in Table 3. It is notable that, under the same reaction conditions, ultrasonic irradiated reactions furnished the corresponding products in shorter reaction times with much higher yields than both room temperature stirring reaction condition and conventional heating condition. In all cases with sonification (200 W), the reactions proceeded smoothly to react with substituted aromatic aldehydes to afford good to excellent yields. However, aromatic aldehydes bearing with electron-withdrawing groups (CF3 and Cl) reacted a little faster than aldehydes bearing with electron-donating substituents (OMe). Moreover, benzimidazolylacetonitrile is also an efficient substrate to undergo with aromatic aldehydes to give the corresponding products within 10 min in 85%–94% yields (Table 3, entries 6–8). To our pleasure, the catalyst SiO2@MNP-A was compatible with 2-indoleacetonitrile, phenylacetonitrile and excellent desired product yields of 85%–95% were obtained when the reaction times were prolonged to 20–60 min (Table 3, entries 9–19). It is noteworthy that all products obtained are E-geometry exclusively and no subsequent Michael adduct is detected because of steric hindrance effect (Scheme 3).
SiO2@MNP-A catalyzed aqueous Knoevenagel condensation of aromatic aldehydes and α-aromatic (heteroaromatic or polyaromatic)-substituted methylene compounds under various reaction conditions.
| Entry | Methylene compound | Aldehyde | Time (min) | Yield (%)a | ||||
| rt | Heat | US | rt | Heat | US | |||
| 1 | Benzothiazole-2-acetonitrile | 4-CF3–C6H4– | 30 | 20 | 1 | 63 | 76 | 90 |
| 2 | Benzothiazole-2-acetonitrile | 2-Cl–C6H4– | 40 | 15 | 2 | 75 | 72 | 83 |
| 3 | Benzothiazole-2-acetonitrile | 4-Cl–C6H4– | 40 | 20 | 2 | 72 | 70 | 87 |
| 4 | Benzothiazole-2-acetonitrile | 4-MeO–C6H4– | 50 | 30 | 8 | 53 | 68 | 92 |
| 5 | Benzothiazole-2-acetonitrile | 2-MeO–C6H4– | 60 | 35 | 10 | 64 | 73 | 91 |
| 6 | Benzimidazolyl acetonitrile | 4-MeO–C6H4– | 40 | 25 | 10 | 72 | 69 | 94 |
| 7 | Benzimidazolyl acetonitrile | 3-MeO–C6H4– | 60 | 30 | 8 | 48 | 66 | 85 |
| 8 | Benzimidazolyl acetonitrile | 3-Cl–C6H4– | 50 | 30 | 6 | 53 | 58 | 85 |
| 9 | 2-Indoleacetonitrile | C6H5– | 120 | 60 | 20 | 36 | 61 | 95 |
| 10 | 2-Indoleacetonitrile | 4–Me–C6H4– | 100 | 60 | 20 | 42 | 59 | 93 |
| 11 | Phenylacetonitrile | C6H5– | 180 | 80 | 30 | 59 | 66 | 95 |
| 12 | Phenylacetonitrile | 4-HO–C6H4– | 180 | 80 | 30 | 63 | 61 | 88 |
| 13 | Phenylacetonitrile | 3-MeO–C6H4– | 150 | 60 | 40 | 63 | 71 | 86 |
| 14 | Phenylacetonitrile | 2-MeO–C6H4– | 280 | 90 | 50 | 65 | 68 | 90 |
| 15 | Phenylacetonitrile | 4-MeO–C6H4– | 180 | 80 | 40 | 63 | 73 | 92 |
| 16 | Phenylacetonitrile | 2-Furyl | 180 | 60 | 35 | 58 | 78 | 92 |
| 17 | Phenylacetonitrile | 1-Naphthyl | 180 | 50 | 35 | 69 | 81 | 95 |
| 18 | 3-Chloro-phenylacetonitrile | 4-MeO–C6H4– | 240 | 100 | 60 | 61 | 67 | 85 |
| 19 | 3-Chloro-phenylacetonitrile | C6H4– | 240 | 100 | 60 | 66 | 65 | 88 |
a Isolated yield based on aldehyde.

(Color online.) Amine functionalized silica coated Fe3O4 nanoparticles (SiO2@MNP-A) catalyzed aqueous Knoevenagel condensation of aromatic aldehydes and α-aromatic substituted methylene compounds.
Encouraged by the exciting results of cyano substituted methylene compounds, we here also attempted to conduct the condensation reaction between non-cyano substituted ketone and benzaldehyde [Scheme 4, eq. (1)]. Gratifying, the reaction of acetylacetone furnished the product (I) in the yield of 79% in water under ultrasonic irradiation at room temperature. Atorvastatin calcium (IV) inhibits 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, which catalyzes the rate-limiting step in cholesterol biosynthesis [24]. As a key intermediate for preparation of Atorvastatin calcium salt (IV), 4-methyl-3-oxo-N-phenyl-2-(phenylmethylene)pentanamide (III) was usually synthesized via the condensation reaction of benzaldehyde and 4-methyl-3-oxo-N-phenylpentanamide (II) in hexane in the presence of the catalytic amount of β-alanine and acetic acid at elevated temperature within 20 h [24c]. The utilization of harmful organic solvent, notorious catalysts and long reaction time more or less brought some problems to develop Atorvastatin calcium in large scale. To our pleasure, SiO2@MNP-A smoothly catalyzed the reaction between benzaldehyde and 4-methyl-3-oxo-N-phenylpentanamide (II) in water under ultrasonic irradiation in 25 min and 87% yield of 4-methyl-3-oxo-N-phenyl-2-(phenylmethylene)pentanamide (III) could be obtained [Scheme 4, eq. (2)]. Thus, this protocol may provide an environmentally friendly alternative for the synthesis of Atorvastatin calcium in industrial scale.
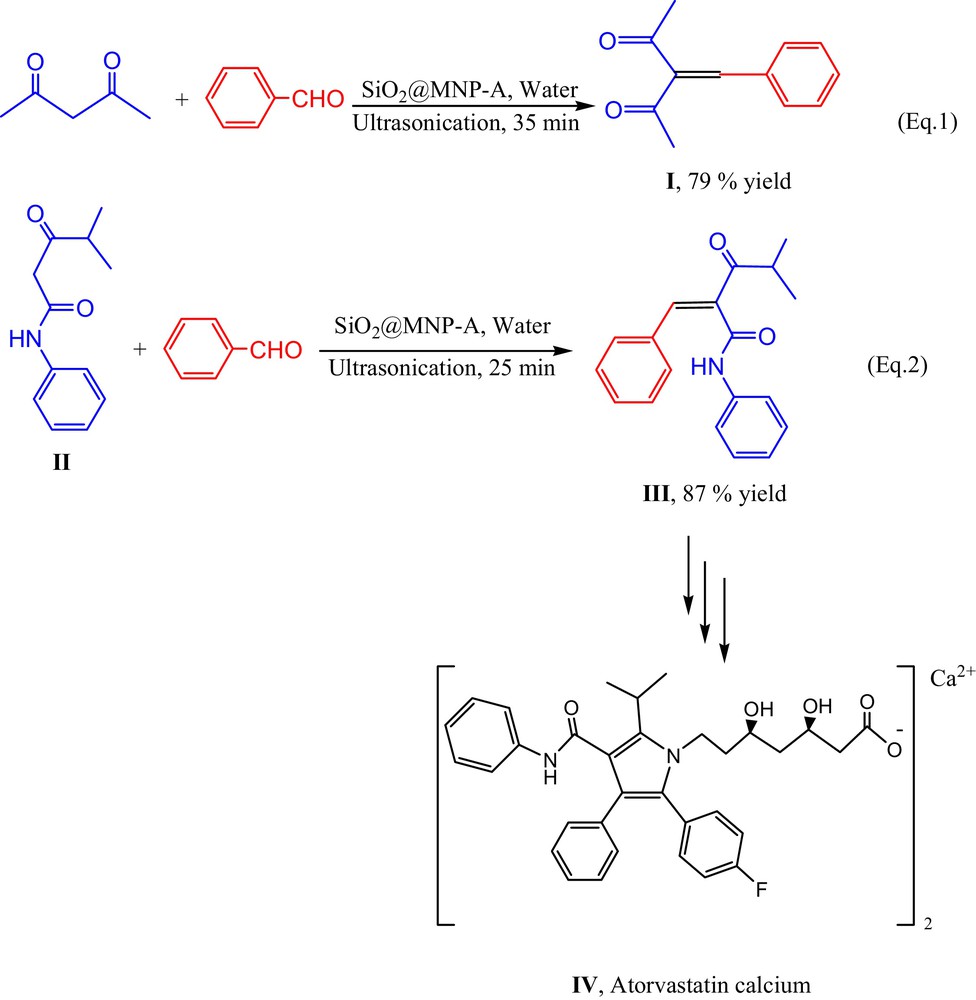
(Color online.) Amine functionalized silica coated Fe3O4 nanoparticles (SiO2@MNP-A) catalyzed aqueous Knoevenagel condensation of non-cyano substituted ketones with benzaldehyde.
In many cases, the solubility of reagents and substrates would determine the reaction efficiency in terms of reaction rate and yield. In this method, the little solubility of benzothiazole-2-acetonitrile in water hampered the reaction proceed further. As demonstrated on Fig. 5, two obvious bands at 217 nm, 254 nm in the three curves were assigned to CC bond vibration of benzene ring (Fig. 5). The absorption magnitude of benzothiazole-2-acetonitrile in water at 217 nm or 254 nm under ultrasonic irradiation is much stronger than that under ambient stirring or that under conventional heating, showing the better solubility of benzothiazole-2-acetonitrile in water under ultrasonic irradiation than that under the remained reaction conditions. The reason for the interesting phenomena may attribute the cavitation in sonochemistry. The chemical and physical effects of ultrasound derive primarily from acoustic cavitation, which includes formations, growth and collapse of the cavity [18]. The implosive collapse of the cavitation forms the bubbles in the liquid phase. Bubble collapse results in high temperature and high pressure within these bubbles, which lead to desired substrate solubility in water as well as the acceleration of the followed reaction rate. A plausible mechanism for the reaction between benzaldehyde and benzothiazole-2-acetonitrile catalyzed by SiO2@MNP-A under ultrasonic irradiation is shown on Fig. 6. With the good aqueous solubility of benzothiazole-2-acetonitrile in hand, the first step is the condensation of the magnetic nanoparticle supported propylamine with benzaldehyde to give imine with good electrophilicity property (Fig. 6A) [25]. After the proton was absorbed by propylamine [26], an activated carbon anion of benzothiazole-2-acetonitrile react with the imine to form the key intermediate B. B is subjected to transfer the proton and electron on the surface of the catalyst to finally afford the condensation product D the recovered catalyst SiO2@MNP-A. The good solubility of benzothiazole-2-acetonitrile in water stimulated by ultrasonic irradiation, the excellent dispersibility of MNPs in reaction solution (Fig. 7a) and high local concentration of substrates around the catalytic sites make the supported catalyst act as a “quasi-homogeneous” promoter which can facilitate the condensation reaction efficiently.
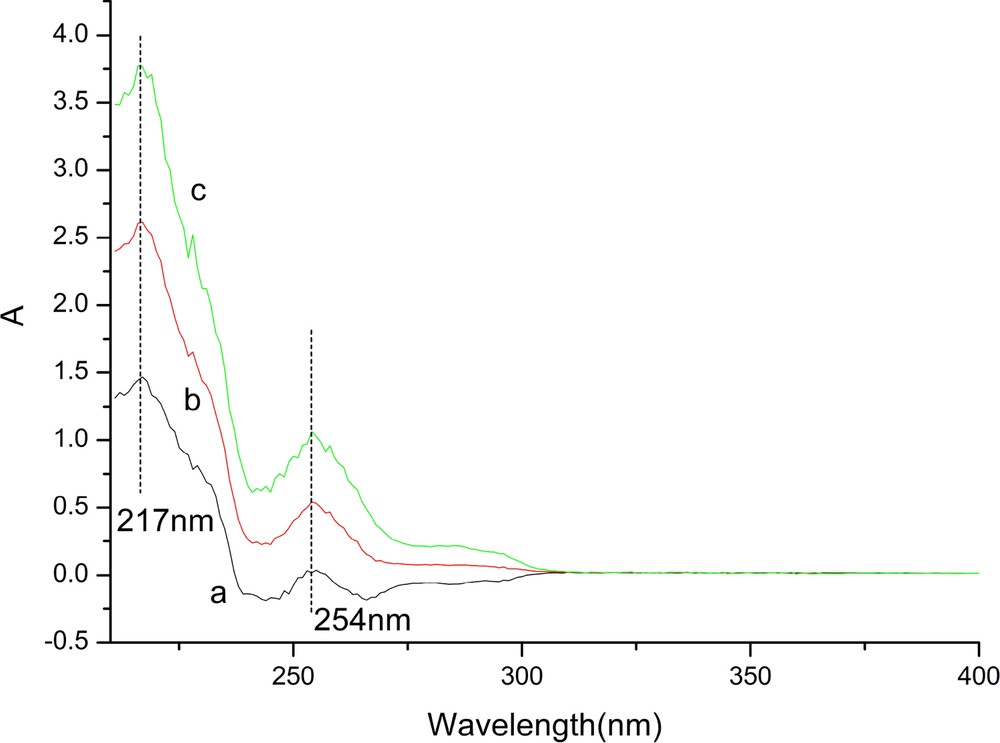
(Color online.) Absorption spectra of benzothiazole-2-acetonitrile in water under various reaction conditions [a: room temperature stirring; b: conventional heating condition (60 °C), c-ultrasonic irradiation (200 W) at room temperature].

(Color online.) Plausible mechanism for the reaction between benzaldehyde and benzothiazole-2-acetonitrile catalyzed by amine functionalized silica coated Fe3O4 nanoparticles (SiO2@MNP-A) under ultrasonic irradiation.

a: amine functionalized silica coated Fe3O4 nanoparticles (SiO2@MNP-A) dispersion in the condensation reaction solution; b: catalyst separation with an external magnet.
For practical applications of heterogeneous catalyst, the recyclability of catalyst is a very important factor. To clarify this issue, catalytic recycling experiments were carried out using the reaction of benzothiazole-2-acetonitrile and benzaldehyde as a model. Upon completion of the reaction, the catalyst SiO2@MNP-A was readily recovered from the reaction mixture with an external magnet (Fig. 7b) and washed with ethanol, subsequently dried at 60 °C for 2 hours under vacuum. The recovered catalyst was reused in the next run and the results are demonstrated on Fig. 8. The results indicate that the catalyst could be simply recovered and reused for 8 times without suffering any significant drop in the catalytic efficiency in terms of reaction yield.
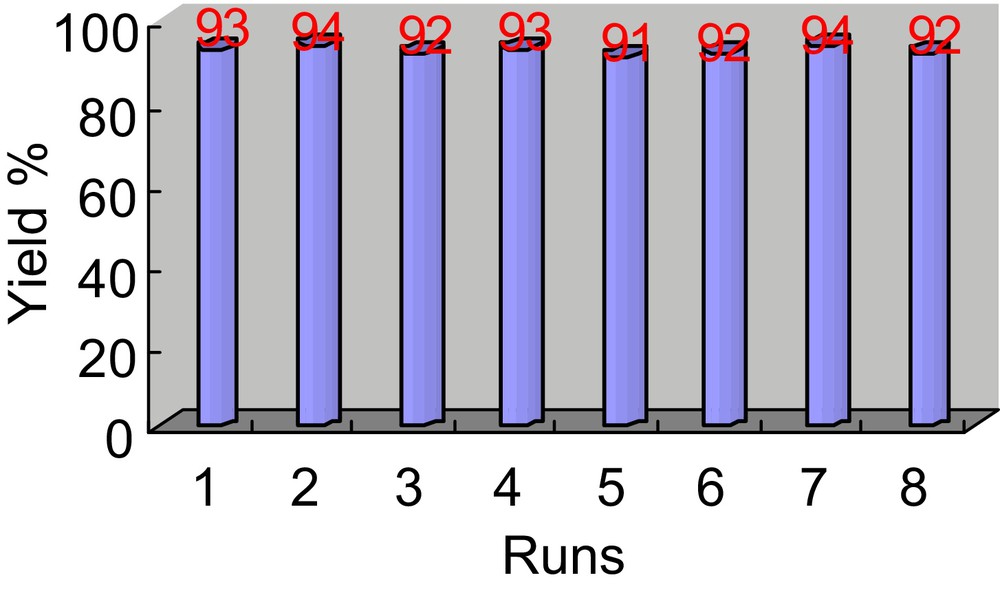
The recyclability of amine functionalized silica coated Fe3O4 nanoparticles (SiO2@MNP-A) in the reaction of benzothiazole-2-acetonitrile and benzaldehyde.
3 Conclusions
In conclusion, we have developed a simple magnetic nanoparticle supported amine (SiO2@MNP-A) and used it as catalyst for ambient Knoevenagel condensation of aromatic aldehydes and α-aromatic (heteroaromatic or polyaromatic)-substituted methylene compounds in water under ultrasonic irradiation. The reaction proceeds smoothly to afford structurally diverse heterocyclic products in good to excellent yields. Moreover, 87% yield of 4-methyl-3-oxo-N-phenyl-2-(phenylmethylene)pentanamide, a key intermediate in the synthesis of Atorvastatin calcium was achieved in the presence of SiO2@MNP-A in water under ultrasonic irradiation at room temperature. The MNP-supported catalyst with high activity because of it “quasi-homogeneous” characteristics, could be readily recovered from reaction mixture by an external magnet, and reused at least 8 times without the significant loss of the catalytic activity. Further preparation and applications of MNP-supported catalysts in organic transformations are underway in our lab.
4 Experimental
4.1 General
All chemicals were purchased from commercial sources and were used without further purification. 1H and 13C NMR were recorded on a Bruker Avance DPX 400 spectrometer at 400 MHz and 100 MHz in CDCl3, respectively. Chemical shifts were reported in parts per million (δ), relative to the internal standard of tetramethylsilane (TMS). IR spectra were recorded on a Nicolet 5700 spectrometer using KBr pellets. Mass spectra were obtained with an automated FININIGAN TSQ Advantage mass spectrometer. Elemental analysis was carried out on a Carlo Erba 1160. TEM was performed with a Phlips Tecnai instrument operating at 40–100 kV. XRD images were obtained a Bruker XRD D8 Advance instrument with Cu Kα radiation. UV–vis concentration evaluation was conducted on a SHIMADZU UV2401-PC spectrometer at room temperature. All sonochemical synthesis were conducted at the ultrasonic instrument (ACE, USA) with the set up of operational frequency (22 kHz). All reactions were monitored by thin layer chromatography (TLC). Flash chromatography was performed on silica gel (100–200 mesh). All condensation products were purified through column chromatography and were characterized by NMR analysis, mass and elemental analysis.
4.2 Preparation of silica coated magnetic nanoparticle (SiO2@MNP)
Magnetic (Fe3O4) nanoparticles were prepared by the coprecipitation [20]. FeCl3·6H2O (8.1 g, 0.03 mmol) and FeCl2·4H2O (4.97 g, 0.025 mmol) were dissolved in distilled water (100 mL). The resulting transparent solution was heated at 85 °C with vigorous mechanically stirring under N2 atmosphere for 1 h. The pH value was then adjusted to 9 using the concentrated aqueous ammonia (25 wt %). After the color of the bulk solution turned to back, the magnetic precipitates were separated and washed several times with deionized water until the pH value of the eluent decreased to 7. The coating of a layer of silica on the surface of the naked Fe3O4 was conducted through sol-gel method [21]. The naked Fe3O4 (1.0 g) was dispersed in ethanol (200 mL) by ultrasonic irradiation. The concentrated NH3·H2O (6 mL) and TEOS (2 mL) were successively added into the solution. With continuous stirring for 24 h at room temperature. The resulting SiO2@Fe3O4 was collected by an external magnet and washed three times with ethanol, followed by drying in vacuum.
4.3 Synthesis of the catalyst SiO2@MNP-A
For 5 min, 0.5 g of Silica coated magnetic nanoparticles was dispersed in dry toluene (30 mL) by ultrasonification. (3-Aminopropyl)triethoxy silane (1.8 g) were added and the reaction mixture was refluxed for 12 h under N2 atmosphere. The supported catalyst was obtained by magnetic force and rinsed with ethanol, dried in a vacuum oven at 60 °C for 12 h. The loading of the catalyst was determined to be 0.75 mmol g−1 by elemental analysis.
4.4 General procedure for the SiO2@MNP-A catalyzed ambient Knoevenagel condensation in water under ultrasonic irradiation
A mixture of benzaldehyde (10 mmol), α-aromatic (heteroaromatic or polyaromatic)-substituted methylene compound (10 mmol), water (20 mL) and SiO2@MNP-A (270 mg) was stirred at room temperature under ultrasonic irradiation. Upon the completion of the reaction (monitored by TLC), the catalyst, separated from the reaction solution by magnet, washed with ethanol and ethyl acetate, followed by drying under vacuum, was reused for subsequent runs. The decanting solution was directly filtrated and the remained residue was purified with recrystallization using ethanol or column chromatography using petroleum ether/ethyl acetate as the eluent. The products were characterized by 1H NMR, 13C NMR, Mass spectra and elemental analysis.
4.4.1 2-(Benzo[d]thiazol-2-yl)-3-(4-methoxyphenyl)acrylonitrile (Table 2, entry 4)
1H NMR (400 MHz, CDCl3): δ 3.90 (s, 3H), 7.01 (d, 2H, J = 6.4 Hz), 7.40–7.53 (m, 2H), 7.89 (d, 1H, J = 6.4 Hz), 8.02–8.07 (m, 3H), 8.18 (s, 1H); 13C NMR (100 MHz, CDCl3): δ 56.8, 102.5, 115.0, 117.3, 121.8, 123.6, 125.4, 125.9, 127.0, 133.0, 135.0, 146.7, 153.8, 163.1, 163.6; ESI-MS: m/z = 293 [M + H]; Anal. Calc. for C17H12ON2S: C 69.85, H 4.13, O 5.47, N 9.58, S 10.97. Found: C 69.78, H 4.18, O 5.52, N 9.53, S 10.99.
4.4.2 2-(1H-indol-2-yl)-3-phenylacrylonitrile (Table 2 entry 9)
1H NMR (400 MHz, CDCl3): δ 7.26–7.33 (m, 2H), 7.40–7.49 (m, 4H), 7.60–7.63 (m, 2H), 7.88 (d, 2H, J = 5.6 Hz), 8.00 (s, 1H), 8.58 (s, 1H); 13C NMR (100 MHz, CDCl3): δ 106.5, 112.4, 113.1, 118.9, 120.0, 121.5, 123.6, 124.4, 125.8, 128.9, 129.1, 129.8, 134.9, 137.3, 138.2; ESI-MS: m/z = 245 [M + H]; Anal. Calc. for C17H12N2: C 83.59, H 4.94, N 11.47. Found: C 83.52, H 4.96, N 11.52.
4.4.3 3-Benzylidene-2,4-pentanedione (Scheme 4, I)
1H NMR (400 MHz, CDCl3) (ppm): 7.47 (s, 1H, C = CH), 7.37 (m, 5H, ArH), 2.40 (s, 3H, CH3), 2.26 (s, 3H, CH3); 13C NMR (100 MHz, CDCl3) (ppm): 200.6, 191.5, 137.8, 134.8, 127.8, 125.6, 124.6, 123.9, 26.6, 21.4.
4.4.4 4-Methyl-3-oxo-N-phenyl-2-(phenylmethylene)pentanamide (Scheme 4, III)
1H NMR (400 MHz, CDCl3): δ 1.21 (d, 6H, J = 6.4 Hz), 3.36 (m, 1H), 7.14–7.18 (m, 1H), 7.32–7.38 (m, 5H), 7.47 (d, 2H, J = 7.6 Hz), 7.56–7.59 (m, 3H), 7.64 (s, 1H); 13C NMR (100 MHz, CDCl3): δ 19.1, 36.7, 120.3, 125.0, 129.0, 129.1, 130.0, 130.1, 133.0, 136.2, 137.4, 140.7, 165.5, 202.6; ESI-MS: m/z = 294 [M + H]; Anal. Calc. for C19H19NO2: C 77.77, H 6.55, N 4.76, O 10.92. Found: C 77.79, H 6.53, N 4.77, O 10.91.
Acknowledgements
We are grateful for the financial supports for this research by the National Natural Science Foundation of China (Grant 21106090 and 21272169), Zhejiang Provincial Natural Science Foundation of China (No. LY12B02004) and we are also grateful for the financial supports for this research by Key Disciplines of Applied Chemistry of Zhejiang Province, Taizhou University.