1 Introduction
Polylactides (PLAs) have been extensively exploited for a wide range of applications, such as sutures, disposal containers, bone fracture fixation devices, controlled release drug carriers, scaffolds, textiles, and tissue engineering [1,2]. In addition, the availability of monomer feedstocks from renewable resources has given PLAs an increasing prominence in the market place. The methods for the synthesis of these polymers now rely on the controlled ring-opening polymerization (ROP) of lactide by well-defined metal initiators, which produce high-molecular weight polymers with good mechanical properties. Also, the stereochemistry of PLAs is a major factor that affects the mechanical properties and biodegradability of these materials [3]. Accordingly, stereoselective ring-opening polymerization of lactides has attracted much interest, leading to a variety of microstructures from the enantiomerically pure monomer, racemic mixture, or meso lactide [4].
An alternative to fossil feedstocks involves using chemicals from renewable resources and also developing new catalytic processes [5,6]. There are strong demands for the development of catalysts and processes that are environmentally clean, safe, stable, and efficient to address today’s urgent need to reduce global environmental pollution and to develop clean and renewable energy resources. In this regard, the past few years have witnessed a renaissance in lithium-based catalysis. Their abundance, availability, and diversity make them useful for a variety of applications of industrial, economic, and environmental importance. For instance, the catalytic activity of lithium derivatives has been described for the ROP of different cyclic monomers, such as caprolactone, cyclic carbonates, and cyclic siloxanes. Lithium complexes are also able to produce PLA from lactide in a controlled manner [7]. For this purpose, several polydentate ligands were employed, such as phenolate scaffolds [8–14], or β-ketoiminate derivatives [15], usually in combination with an external alcohol. Despite numerous reports on ROP of l-lactide, very few studies are available in the literature that describe the stereoselective ROP of rac-lactide by metal alkali complexes [16]. Kasperczyk et al. first reported that in the presence of tBuOLi heterotactic-enriched PLA (Pr = 0.75) was obtained, albeit low conversion of rac-lactide was reported [17]. Since then, only iminophenolate lithium complexes could also produce PLA with similar heterotacticity [18].
Therefore, we were interested in investigating the ROP of rac-lactide with other lithium-based phenolate systems. As salan and salen ligands are interesting platforms which have not so far been evaluated for the ROP of lactide in the presence of lithium, our attention was drawn to the known salan [19] and salen ligands 1 and 2, the new chiral salen derivative 3 and the new imidazolinium salts 4 and 5 [tridentate N-heterocyclic carbenes (NHC) precursors] (Fig. 1). We report herein the synthesis of ligands 3–5 and the activity of the lithium complexes prepared in situ in the presence of ligands 1-5 for the heterotactic ROP of rac-LA.
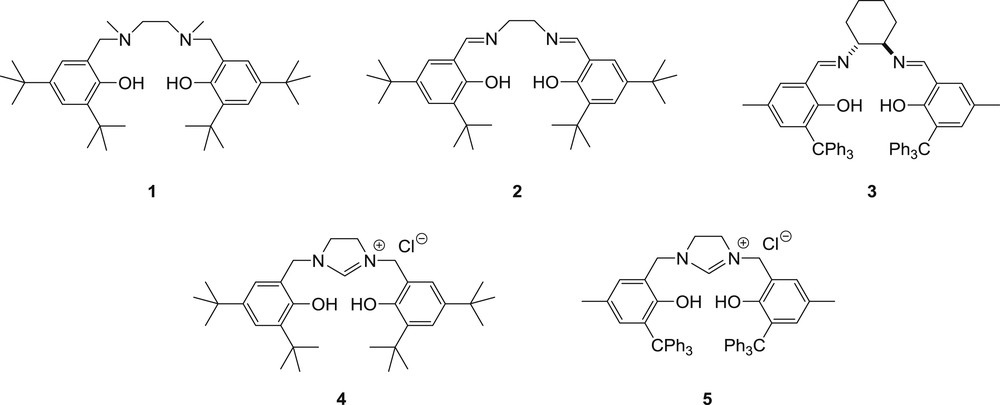
Ligands screened in this study.
2 Results and discussion
Ligand 3 was easily formed by condensation of 1,2-diaminocyclohexane and 2-hydroxy-5-methyl-3-tritylbenzaldehyde in excellent yield (94%). The synthesis of the new imidazolinium salts 4 and 5 was also carried out by a conventional procedure. Symmetrical imidazolinium salts are often prepared by cyclisation of a diamine with triethyl orthoformate in the presence of an appropriate salt [20]. Therefore, for the preparation of 4 and 5, cyclisation of the corresponding diamines 4a and 5a was performed at 80 °C for 20 hours in a chloroform/triethyl orthoformate (1:2) mixture with a catalytic amount of acetic acid. The presence of ammonium chloride was required to form the imidazolinium chloride salts. Ligand 4 was obtained in a satisfying yield of 65%, while 5 was produced with a lower yield of 43%, probably due to the presence of a bulkier ortho-substituent (Scheme 1).
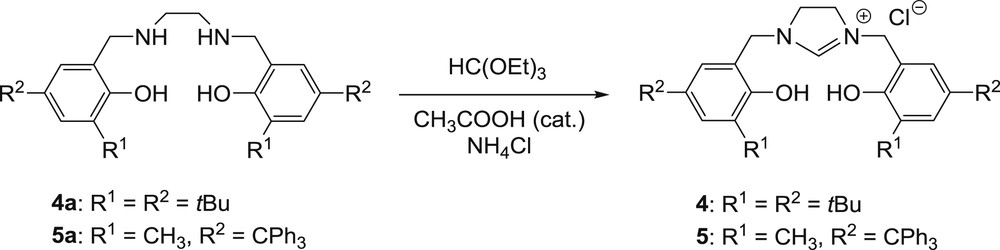
Synthesis of imidazolinium chloride salts 4 and 5.
The complexes were prepared in situ by addition of 2 equiv of nBuLi in THF at low temperature. Single crystals suitable for X-ray structure determination of (1-Li2)2 were obtained from slow cooling of a warm THF/hexane solution. The molecular structure of (1-Li2)2 illustrates an aggregation mode in the solid state with four lithium atoms (Fig. 2). In the solid state, compound (1-Li2)2 possesses a ring-laddering conformation. This architectural structure of alkali metal chemistry has been discussed extensively in the literature [21,22]. We hypothesized that similar complexes with ligands 2 and 3 were obtained. We could also visualize the deprotonation of the phenols of the salen ligand 3 in the presence of 2 equiv of nBuLi by 1H NMR (see Fig. S9). Regarding the NHC precursors 4 and 5, no crystal structure was obtained but the 1H NMR spectra of the in situ generated complexes showed that the imidazolinium proton was deprotonated in the reaction conditions.
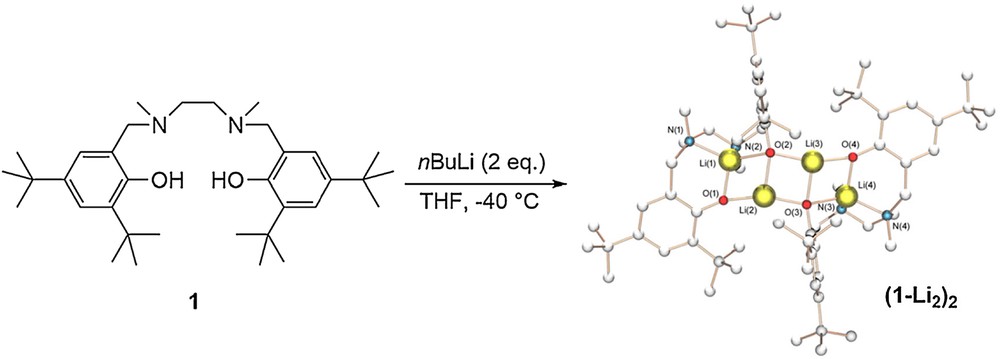
(Color online.) Synthesis and solid-state structure of complex (1-Li2)2. Selected bond lengths (Å): N(2)–Li(1) 2.057(4), O(2)–Li(3) 1.934(4), O(2)–Li(2) 1.947(4), O(2)–Li(1) 1.980(4), Li(2)–O(1) 1.858(4), Li(2)–O(3) 1.934(4), N(1)–Li(1) 2.005(4), O(1)–Li(1) 1.866(4), N(4)–Li(4) 2.006(4), O(4)–Li(4) 1.850(4), O(4)–Li(3) 1.855(4), Li(4)–O(3) 1.975(4), Li(4)–N(3) 2.055(4), O(3)–Li(3) 1.940(4).
3 ROP of rac-LA with bimetallic complexes prepared in situ
As we recently reported the heterotactic ROP of rac-lactide using hetero-bimetallic Mg–Li and Zn–Li complexes in combination with bisphenol ligands, we decided to use the same reaction conditions in this study [23]. Therefore, the polymerization tests were performed in a THF/toluene (1:5) solvent mixture with 1 equiv of neopentyl alcohol (NpOH) and 50 equiv of rac-LA (Scheme 2). The bisphenol systems were first screened at room temperature. The resulting lithium-based complexes were active as they led to quantitative conversion of rac-LA in less than 15 minutes [the reaction times were not fully optimized therefore the turnover frequencies (TOF) up to 600 h−1 are underestimated] and the resultant polymers reveal experimental number average molecular masses (Mn) close to the theoretical ones (Table 1, entries 1–3, 6, 9). Narrow molecular weight distributions were obtained (Mw/Mn between 1.32 and 1.41) except with ligand 1.
Synthesis of heterotactic PLA.
Ring-opening polymerization of rac-lactide by Li complexes prepared in situ from ligands 1 to 5 in the presence of 1 equiv of neopentyl alcohol.a
| Entry | Ligand | T (°C) | t (min) | Conv.b,c (%) | Mn, expd (g/mol) | Mw/Mnd | Pre | TOF (h−1) |
| 1 | 1 | 20 | 15 | 100 | 7300 | 1.60 | 0.50 | 200 |
| 2 | 2 | 20 | 25 | 100 | 6400 | 1.34 | 0.58 | 120 |
| 3 | 3 | 20 | 15 | 100 | 6800 | 1.38 | 0.78 | 200 |
| 4 | 3 | 0 | 25 | 94 | 7500 | 1.37 | 0.81 | 113 |
| 5 | 3 | –30 | 55 | 92 | 8400 | 1.31 | 0.86 | 50 |
| 6 | 4 | 20 | 5 | 100 | 8000 | 1.41 | 0.66 | 600 |
| 7 | 4 | 0 | 30 | 100 | 7400 | 1.47 | 0.80 | 100 |
| 8 | 4 | –30 | 100 | 98 | 6500 | 1.51 | 0.78 | 30 |
| 9 | 5 | 20 | 10 | 100 | 7400 | 1.32 | 0.81 | 300 |
| 10 | 5 | 0 | 30 | 100 | 7600 | 1.54 | 0.80 | 100 |
| 11 | 5 | –30 | 60 | 81 | 7500 | 1.50 | 0.86 | 41 |
| 12f | 3 | 20 | 8 | 100 | 15,840 | 1.80 | 0.78 | 750 |
a [LA] = 1 M in a THF/toluene (1:5) mixture with 50 equiv of LA (except for Entry 12).
b Conversion calculated using 1H NMR.
c Reaction time not optimized for total conversion.
d Mn exp and Mw/Mn determined by SEC-RI using polystyrene standards in THF. Values corrected by Mark–Houwink factors (0.58).
e Pr is the probability of racemic linkages between monomer units and is determined from the methine region of the homonuclear CH3-decoupled 1H NMR spectrum at 25 °C.
f Reaction run with 100 equiv of LA.
An important goal of this work was to study the asymmetric incorporation of rac-lactide into the polymer backbone. For this purpose, 1H NMR spectroscopy is applicable for determination of the stereochemistry of the PLAs by inspection of the methine region of homonuclear decoupled 1H NMR spectra of the polymers (see Fig. S10). Microstructural analysis of the different PLAs formed from rac-LA revealed that at room temperature the lithium-based systems 3-Li2/NpOH, 4-Li2/NpOH and 5-Li2/NpOH exert a significant influence on the tacticity of the polymer formed, as the microstructure is heterotactic (entries 3, 6 and 9). By contrast, complexes 1-Li2/NpOH and 2-Li2/NpOH produced atactic PLA (entries 1–2). As already observed for other ROP systems, this decrease in tacticity is consistent with previous observations that ligand substituents are primordial for stereochemical control through a chain-end control mechanism [24–26]. The activity and selectivity of the most promising complexes were then investigated at lower temperature. Satisfyingly, catalysts 3-Li2/NpOH, 4-Li2/NpOH and 5-Li2/NpOH were still active at –30 °C with TOF comprised between 30 and 50 h−1. At 0 °C, the heterotacticity was similar to those at room temperature, and was increased to 86% at –30 °C with ligands 3 and 5 bearing trityl ortho-susbtituents (entries 5 and 11). This Pr value is, to our knowledge, the highest observed for alkali metal complexes. The ratio [LA]/[I] was increased to 100 with complex 3-Li2/NpOH and the polymerization reached 100% conversion in 8 minutes at room temperature (entry 12). Experimental Mn value was in accordance to theoretical one (15,840 vs 14,400 g/mol) but a larger molecular weight distribution was observed (Mw/Mn = 1.80). A Pr value of 0.78 was achieved, similar to the one obtained in the presence of 50 equiv of rac-LA. Finally, sodium and potassium complexes using the sterically hindered trityl salen ligand 3 were also prepared in situ in the presence of NaN(SiMe3)2 and tBuOK, respectively. These salts were then evaluated for the ROP of rac-LA with [LA]/[I] = 50 at room temperature. The polymerization was controlled with both complexes (Mn exp = 6200–7200 g/mol; Mw/Mn = 1.38–1.41) but atactic polymers were produced.
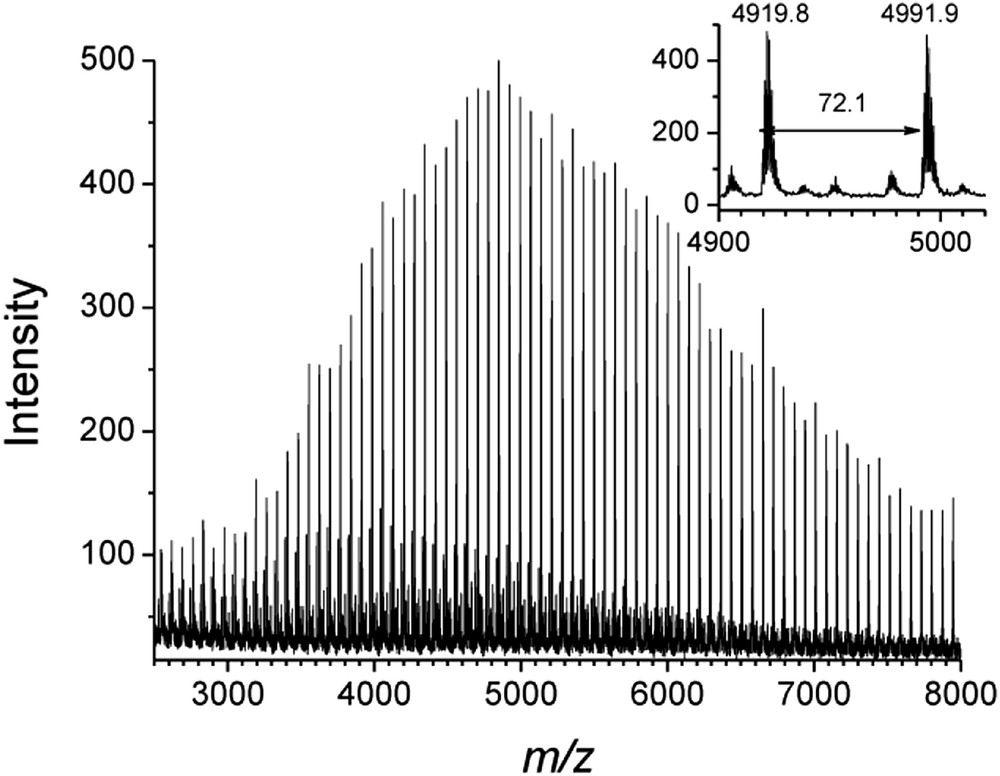
PLA oligomers produced with the lithium system 3-Li2/NpOH were analyzed by MALDI-TOF mass spectrometry (Fig. 3). End-group analysis revealed that the polymer chains were end-capped by one neopentyl ester group and one hydroxyl group. As it is generally accepted, these MALDI-TOF mass spectra confirmed that the ring-opening of LA by the lithium complex 3-Li2/NpOH proceeds by a monomer activated mechanism, where the lactide monomer is activated by coordination to the Li+ center, and consequently NpOH can attack the carbonyl group to initiate the ROP reaction [27]. Also, the spectra exhibit a series of peaks separated by 72 m/z, half that of the LA monomer (144 m/z) (Fig. 3). The generation of odd-numbered PLAs can be accounted for some transesterification reaction. A minor series of peaks was also observed on the MALDI-TOF spectra, which were attributed to cyclic polyester.
4 Conclusion
In this study, we have disclosed the catalytic activity of several lithium-based systems in combination with a primary alcohol in the ROP of lactide. These bisphenolate catalytic systems have been shown to feature high performances for the polymerization of lactide, affording PLAs with a high degree of control in terms of molecular weight, molecular weight distribution, and tacticity. These lithium-based systems are therefore promising in order to create more versatile, active and selective systems for the ROP of bio-sourced monomers. In this regard, variations of the catalyst precursor and the ligand, as well as attempts to isolate the active catalysts are currently ongoing in our laboratories, in the goal of improving the stereoselectivity of the methodology.
5 Experimental
5.1 General considerations
All reactions were carried out under an argon atmosphere using Schlenk techniques or a glove box (< 1 ppm O2, < 2 ppm H2O). Tetrahydrofuran and toluene were dried with a solvent purification system (PureSolv, Innovative Technology, Inc.) and degassed by freeze pump thaw cycles prior to use. THF-d8 is freshly distilled from benzophenone/sodium prior to use. Purification of rac-lactide includes repeated recrystallization from isopropanol or toluene and sublimation. Neopentyl alcohol was purified by sublimation. Paraformaldehyde was used as received from Sigma–Aldrich. 1,2-diaminocyclohexane and xylene were used as received from Alfa Aesar. Acetic acid, ammonium chloride, triethyl orthoformate were used as received from Acros. Chloroform was used as received from VWR Prolabo. N,N′-bis(3,5-di-tert-butyl-2-hydroxybenzyl)-N,N′-bis(methylene)-1,2-ethanediamine 1 [28]; (S,S)-(+)-N,N′-bis(3,5-di-tert-butyl-salicylidene)-1,2-ethanediamine 2 [29], 2-hydroxy-5-methyl-3-tritylbenzaldehyde [30], N,N′-bis(3,5-di-tert-butyl-2-hydroxybenzyl)ethylenediamine 4a [31] were prepared by literature methods. NMR spectra were recorded on a Bruker Avance 300 spectrometer or a Bruker Avance 400 spectrometer (at the “École nationale supérieure de chimie de Paris”). Chemical shifts are reported in ppm and coupling constants J in hertz. NMR spectra were calibrated using residual 1H and 13C resonances of deuterated solvents (1H, δ 7.26 pour CDCl3, δ 3.58 for THF-d8; 13C{1H}, δ 67.21 for THF-d8). MALDI-TOF mass spectra were run with Perseptive Voyager DE-STR MALDI-TOF spectrometer (Perseptive Biosystems, Framingham, MA, USA). trans-2-[3-(4-tert-Butylphenyl)-2-propenylidene] malonitrile (DCTB) used as the matrix for MALDI-TOF MS, was of highest grade available (used without further purification) and was purchased from Sigma–Aldrich. Size exclusion chromatography (SEC) of polymers was performed in THF at 35 °C using an Agilent 1260 Infinity Series GPC [ResiPore 3 μm, 300 × 7.5 mm, 1.0 mL/min, UV (250 nm) and RI (PL-GPC 220) detectors]. The number average molecular masses (Mn) and polydispersity index (Mw/Mn) of the polymers were calculated with reference to universal calibration vs. polystyrene standards (limits Mw = 200 to 400,000 g/mol).
5.2 (R,R)-(–)-N,N′-bis(3,5-di-triphenylmethylsalicylidene)-1,2-cyclohexanediamine 3
A mixture of 1,2-diaminocyclohexane (10.7 mmol, 0.08 g) and 2-hydroxy-5-methyl-3-tritylbenzaldehyde (1.4 mmol, 0.5 g) in 10 mL of dichloromethane was stirred under reflux overnight. All volatiles were removed via rotary evaporator. The product (3) was then was purified by recrystallization in ethanol (yield 94%). 1H NMR (CDCl3, 300 MHz): δ 7,92 (2H, s, CHN); 7.27–7.18 (30H, m, Ar); 7,07 (2H, m, Ar); 6.85 (2H, m, Ar); 3.03 (2H, m, Cy); 2,28 (6H, s, Me); 1.78–1.74 (4H, m, Cy); 1.52 (2H, m, Cy); 1.30 (2H, m, Cy). 13C NMR (CDCl3, 75 MHz): δ 164,7 (NCHAr); 157.8 (PhOH); 145.7 (CPh3); 134.2 (PhOH); 131.0 (CPh3 + PhOH masked); 127.1 (CPh3); 125.9 (PhOH); 125.5 (CPh3); 118.4 (PhOH); 72.8 (Cy); 63,1 (CPh3); 32.8 (Cy); 24.2 (Cy); 20.8 (Me).
5.3 1,3-bis(3,5-di-tert-butyl-2-hydroxybenzyl)-4,5-dihydro-1H-imidazolinium chloride 4
N,N′-bis(3,5-di-tert-butyl-2-hydroxybenzyl)ethylenediamine (9,25 mmol, 4,60 g) was dissolved under argon in 50 mL of chloroform before triethyl orthoformate (100 mL, large excess) and ammonium chloride (11,21 mmol, 600 mg) were added. The reaction was carried out in the presence of catalytic amount of acetic acid. The reaction mixture was stirred for 20 hours at 80 °C. After cooling to the room temperature, solvents were evaporated under vacuum. The residual solid was purified by column chromatography (the product eluted by CH2Cl2/EtOH 5:1) to give 4 as a pale solid (3,27 g, 65%). IR (cm−1) 1650, 2965, 3161, 3630. 1H NMR (CDCl3, 300 MHz): δ 8.55 (s, 1H, CHim), 7.31 (d, 2H, J = 3,0 Hz, CHAr), 6.91 (d, 2H, J = 3,0 Hz, CHAr), 5.05 (s, 4H, CH2), 3.83 (s, 4H, CH2), 1.41 (s, 18H, t-Bu), 1.25 (s, 18H, t-Bu). 13C NMR (CDCl3, 75 MHz): δ 157.5 (CHim), 152.2 (CAr), 143.6 (CAr), 140.1 (CAr), 140,0 (CHAr), 125.4 (CHAr), 125.3 (CAr), 121.1 (Car), 49.9 (CH2), 48.1 (CH2), 35.1 (Ct−Bu), 34.2 (Ct−Bu), 31.6 (t-Bu), 30.1 (t-Bu). Anal. Calcd. for C33H51ClN2O2: C, 72.96; H, 9.46, N, 5.16. Found C, 71.45; H, 9.50, N, 5.07. HRMS, m/z: (M)+ calcd for C33H51ClN2O2: 507.39476, found: 507.39451.
5.4 N,N′-bis(3-methyl-5-trityl-2-hydroxybenzyl)ethylendiamine 5a
A mixture of ethanediamine (1 equiv, 4.6 mmol, 0.31 mL) and 2-hydroxy-5-methyl-3-tritylbenzaldehyde (2 equiv, 9,3 mmol, 3.51 g) in 200 mL of dichloromethane was stirred under reflux during 4 hours. Then, the mixture was cooled with an ice bath before adding 40 mL of EtOH and sodium borohydride (3 g, large excess). The mixture was then quenched with NH4Cl. After extraction with CH2Cl2/NH4Cl, the compound was dried over MgSO4. Solvents were evaporated under vacuum. The residual solid was purified by crystallization in CH2Cl2/pentane to give 5a as a white solid (2.52 g, 69%). 1H NMR (CDCl3, 300 MHz): δ 7.30–7.15 (30H, m, Ar); 6.92 (2H, m, Ar); 6.77 (2H, m, Ar); 3.77 (4H, s, CH2N); 2.21 (6H, s, Me); 2.13 (2H, s, NCH2CH2N). 13C NMR (CDCl3, 75 MHz): δ 154.0 (PhOH); 146.1 (CPh3); 134.1 (PhOH); 131.2 (CPh3); 130.6 (PhOH); 128.6 (PhOH); 127.0 (CPh3); 126.7 (PhOH); 125.4 (CPh3); 122.1 (PhOH); 63.2 (CPh3); 52.3 (NCH2CH2N); 46.3 (NHCH2Ar); 28.4 (Cy); 21,0 (Me).
5.5 1,3-bis(2-hydroxy-5-methyl-3-tritylbenzyl)-4,5-dihydro-1H-imidazolinium chloride 5
N,N′-bis(3-methyl-5-trityl-2-hydroxybenzyl)ethylenediamine 5a (2,54 mmol, 2,00 g) was dissolved under argon in 50 mL of chloroform before triethyl orthoformate (100 mL large excess) and ammonium chloride (3,74 mmol, 200 mg) were added. The reaction was carried out in the presence of a catalytic amount of acetic acid. The reaction mixture was stirred for 20 hours at 80 °C. After cooling to room temperature, the solvents were evaporated under vacuum. The residual solid was purified by column chromatography (the product was eluted by CH2Cl2/EtOH) to give 5 as a pale solid (0,80 g, 43%). IR (cm−1) 1647, 2911, 3030. 1H NMR (CDCl3, 300 MHz): δ 9.24 (s, 1H, CHim), 7.9–7.06 (m, 32H, CHAr), 6.92 (s, 2H, CHAr), 4.61 (s, 4H, CH2), 3.28 (s, 4H, CH2), 2.22 (s, 6H, CH3Ar). 13C NMR (CDCl3, 75 MHz): δ 158.5 (CHim), 150.7 (CAr), 144.0 (CAr), 134.3 (CAr), 132,3 (CHAr), 131.3 (CHAr), 130.9 (Car), 130.1 (CAr), 128.0 (Car), 126.9 (Car), 121.2 (Car), 62.8 (CH2), 48.0 (CH2), 47.5 (CH2), 20.9 (CH3). Anal. Calcd. for C57H51ClN2O2: C, 82.34; H, 6.18, N, 3.37. Found C, 78.98; H, 6.03, N, 3.34. HRMS, m/z: (M)+ calcd for C57H51ClN2O2: 795.39505, found: 795.39451.
5.6 In situ preparation of L-Li2 catalyst
The appropriate ligand (0.03 mmol) was dissolved in anhydrous THF (0.5 mL) and then placed 1 hour in the fridge. n-BuLi (2.5 M in hexane, 0.06 mmol, 24 μL) was added dropwise in the Schlenk tube. The mixture was stirred at room temperature for 1 hour, yielding complex L-Li2 in situ (100%), which was engaged in the next step (polymerization) without further purification.
5.7 In situ preparation of 3-Na2 catalyst
Ligand 3 (20.9 mg, 0.025 mmol) was dissolved in anhydrous THF (0.25 mL) and then placed during 1 hour in the fridge. Na[N(SiMe3)2] (9.2 mg, 0.05 mmol) was added in the Schlenk tube. The mixture was stirred at room temperature for 1 hour, then the Schlenk tube was placed during 1 hour in the fridge, yielding complex 3-Na2 in situ, which was engaged in the next step (polymerization) without further purification.
5.8 In situ preparation of 3-K2 catalyst
Ligand 3 (20.9 mg, 0.025 mmol) was dissolved in anhydrous THF (0.25 mL) and then placed 1 hour in the fridge. tBuOK (5.6 mg, 0.05 mmol) was added in the Schlenk tube. The mixture was stirred at room temperature for 1 hour, then the Schlenk tube was placed for 1 hour in the fridge, yielding complex 3-K2 in situ, which was engaged in the next step (polymerization) without further purification.
5.9 Polymerization of rac-lactide with in situ prepared catalyst
Neopentyl alcohol (8.8 mg, 0.1 mmol) and toluene (3 mL) were added to the solution of the in situ complex previously obtained (0.75 mL, 0.1 mmol). The Schlenk tube was placed for 1 hour in the fridge. Then rac-lactide (720 mg, 5 mmol, 50 equiv) was added and the mixture was stirred at room temperature for the appropriate time before being quenched. The solvents were removed under vacuum and the polymer was precipitated in an Et2O/pentane (2:1) mixture. Finally, the polymer was dried under vacuum.
Acknowledgments
CNRS, ENSCP, the “Fondation Pierre-Gilles-de-Gennes”, and the French Ministry of Research and Higher Education are thanked for financial support of this work. The authors would like to thank Purac for a generous loan of rac-lactide. CMT is grateful to the “Institut universitaire de France” (IUF).