

1 Introduction
The debilitating effects of spinal cord injuries (SCI) are not only induced by direct disruption of spinal cord ascending and descending pathways, but also by post-traumatic complications, known as the second injury, which in part encompasses ischemia, hematotoxicity, excitotoxicity, inflammatory reactions and release of molecules inhibiting axonal regrowth [1]. As a result, both acute and chronic therapeutic interventions have been designed to potentiate the limited spontaneous recovery that is sometimes observed in SCI patients. They usually consist in limiting secondary injury, promoting plasticity or axonal regrowth through rehabilitation, cell transplantation [2], or molecule administrations (e.g. neurotrophic factors and antagonists for neurite growth inhibitors) [3]. Although promising results have been obtained in animal studies, no efficient clinical therapy, except rehabilitation, is yet available [4]. This poor translation from animal research evidence to effective clinical treatments could be, in part, caused by the shortcomings of cell transplantation and molecule administration. Indeed, it was noted that a majority of cells undergoes apoptosis after implantation due to the hostility of the lesion site environment and the lack of cell adhesion [5]. In addition, direct molecule injection leads to uncontrolled molecule release kinetics [6].
An alternative strategy is to fill the spinal cord cavity with a polymer biomaterial in order to reduce the glial scar, which is known to impede axonal regrowth, and to increase axonal regeneration by supplying a substrate for growth support [7]. However, to efficiently repair the spinal cord the characteristics of implanted biomaterials should properly match the ones of the nervous tissue’s extracellular matrix (ECM) as it was shown that the ECM played a key role in the migration, the morphology, the phenotype and the survival of cells [8]. A recent study even demonstrated that the implantation of an acellular spinal cord scaffold, obtained through a simple series of detergent, induced functional recovery in hemisected rats [9]. In that context, hydrogels, which are three-dimensional networks formed by hydrophilic cross-linked polymers swollen in water, seem to be well suited for both spinal cord and brain repair [10]. Hydrogels can easily be cast into various shapes in order to properly fill the lesion site or to present pores and microchannels [11]. Moreover, by adjusting the synthesis process it is possible to obtain hydrogels with modulus of elasticity that closely fits the soft mechanical properties of the nervous tissue [12]. Hydrogel mechanical properties are not only known to profoundly affect the neuronal morphology such as neurite branching [13] but also to regulate the self-renewal and the differentiation of the neural stem cell (NSC) [14]. In fact, hydrogels are well suited to be used as cell carriers as it was shown that cells delivered via a biocompatible scaffold presented high survival rates than transplanted cells through simple injection [5].
To date, various polymers from both natural and synthetic origins have already induced therapeutic effects in different animal models [15–17]. Nevertheless, synthetic hydrogels are particularly attractive as they present a low batch-to-batch variability and can be manipulated to have a wider range of physico-chemical properties than natural ones. Additionally, implantation of synthetic materials avoids disease transmission and reduces allergenic and immunogenicity risks [16, 18]. Synthetic hydrogels used in spinal cord repair principally consist of polymethacrylates/methacrylamides and polyethers whose cross-linking is based on different chemistry approaches (e.g. radical polymerization and click reactions). They have been extensively studied and brought promising results. Indeed, functional improvement, tissue recovery and axonal regeneration have been observed after implantation of poly[N-(2-hydroxypropyl)methacrylamide] (PHPMA) [19], poly(2-hydroxyethyl methacrylate) (PHEMA) [20], and poly(ethylene glycol) (PEG) hydrogels, respectively [21]. Although hydrogel implantation induces therapeutic effects by itself, synergistic outcomes are generally observed with cell-seeded scaffolds [22]. Indeed, Hejčl et al. only observed significant functional improvement after the implantation of the mesenchymal stem cell (MSC)-seeded PHPMA hydrogel in a chronic SCI model [23]. Other authors studied injectable in situ-forming hydrogels in order to limit the surgical invasiveness of conventional implantation [16].
Many well-written reviews already discussed the therapeutic effects induced by the implantation of biomaterials after a spinal cord injury [3, 15–17]. In those reviews, the biomaterials were either classified depending on their nature or their characteristics and both advantages and drawbacks of each biomaterial were debated. In particular, the review of Straley et al. in 2010 has thoroughly listed the desired characteristics of hydrogels for use in spinal cord repair. Besides complex questions about mechanical compliance, adapted porosity/topography, injectability and cell encapsulation, the authors pointed out the crucial importance of cell adhesion, biomolecule delivery and degradability in the design of hydrogel scaffolds. In this short review, we precisely focus on the chemical strategies recently developed to obtain synthetic hydrogels displaying these three key features for use in spinal cord repair, namely (i) improved surface biofunctionality regarding cell adhesion, (ii) drug delivery ability, and (iii) degradability properties so as to avoid chronic complications [3, 18]. We included both in vivo and in vitro studies on tissue regeneration obtained with these scaffolds. Although each section of this review deals with definite characteristics, it is important to keep in mind that they are all intertwined.
2 Bioadhesion promoting strategies
In spite of exhibiting the interesting above-mentioned features, synthetic polymer-based biomaterials do not generally present inherent surface properties that can provide a suitable environment for biological processes (primarily cell adhesion). Therefore, many efforts have been devoted to modify the bioinert surface of these biomaterials by ECM proteins (e.g. laminin, fibronectin, and collagen), which have the specific advantage of possessing cell-adhesive or signaling domains. Such strategy is not only known to increase tissue integration and viability of transplanted cells but also to improve axonal regeneration. Indeed, after a complete spinal cord transection, a higher number of regenerating axons were observed in fibrin-filled poly(2-hydroxyethyl methacrylate-co-methyl methacrylate) (PHEMA-MMA) implanted channels than in unfilled channels [24]. Similarly, an increase in neurite growth was noticed in vitro on the PEG hydrogel coated with fibronectin (i.e. an ECM glycoprotein) [25]. However, ECM-derived proteins present multiple limitations such as disease transmission, risks of degradation and inadequate adsorption leading to poor efficiency for cell adhesion. Indeed, full-length ECM proteins might undergo conformational changes that could induce a foreign body response or result in the masking of the binding domains needed to promote the cell adhesion [26]. As a result, studies are now being increasingly focused on polymer modification with only the biologically active peptide sequences derived from these ECM molecules, such as the well known arginine-glycine-aspartic acid (RGD) or isoleucine-lysine-valine-alanine-valine (IKVAV) [27], that are chemically well defined and much easier to prepare (automated peptide synthesis) and handle. Nevertheless, a recent strategy showed the possibility to specifically immobilize full-length proteins so as to retain their active conformation. This strategy involved polymer surface modification with streptavidin, through the use of a photo-cross-linkable diazirine agent, and further binding with the biotinylated protein. Such immobilized protein induced a superior neurite outgrowth than its analog that was simply adsorbed on the polymer in a non-controlled manner [28].
In the context of long-term implants, such as in spinal cord repair, peptides of interest are preferentially attached to biomaterials through covalent bindings in order to limit peptide removal over time [29]. Numerous studies regarding bioactive peptide functionalization have been performed on methacrylamide/methacrylate-based hydrogels, through various approaches. For example, a PHPMA-RGD hydrogel was synthetized by radical copolymerization of HPMA with a methacrylamide-terminated GGRGD peptide, obtained from reaction of N-terminal amine of the GGRGD peptide with methacryloyl chloride [30, 31]. Ethylene dimethacrylate was also used as a comonomer during the polymerization to induce chain cross-linking, as well as solid porogen (sodium chloride) to impart porosity. This specific hydrogel then was seeded with MSC before being implanted into a hemisected thoracic rat spinal cord. As expected, the PHPMA-RGD hydrogel presented a better combination of properties for tissue regeneration than the non-functionalized PHPMA hydrogel (Fig. 1A).
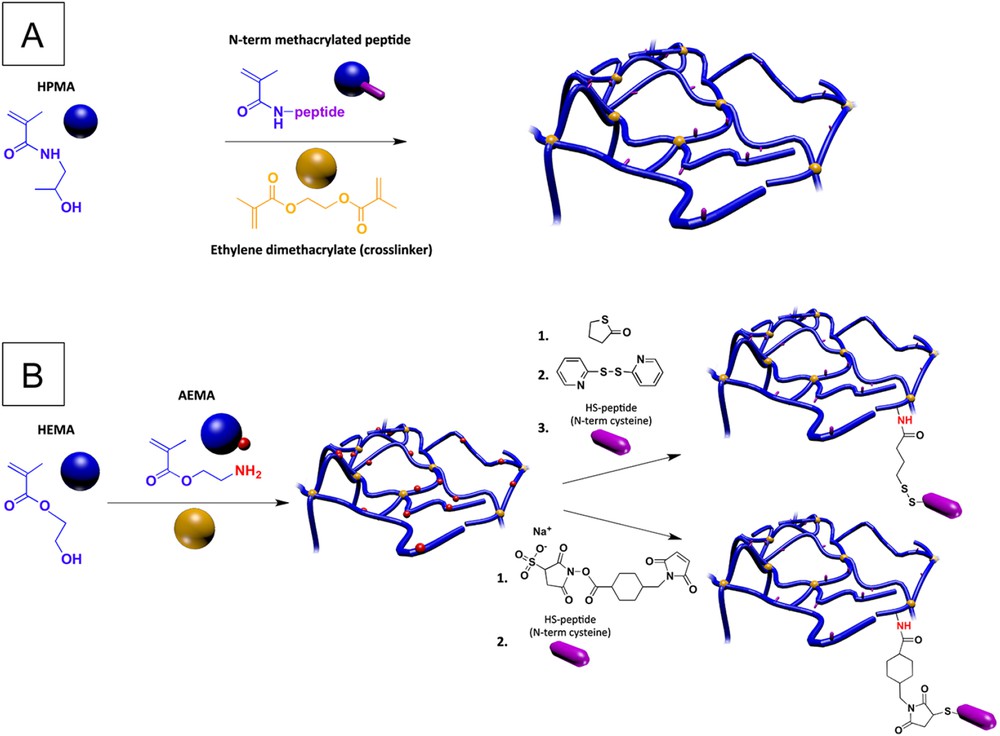
Chemical approaches for PHPMA/PHEMA hydrogel functionalization with cell adhesion peptides; (A) use of an N-terminus methacrylated peptide (RGD based sequence) as a comonomer for polymerization; (B) post-polymerization method for coupling of thiol-terminated peptides (IKVAV and YIGSR based sequences), based on bifunctional dithiopyridine (top pathway) or sulfo-SMCC (bottom pathway) linkers.
The binding motif can also be incorporated into a post-polymerization approach, as reported for PHEMA-based hydrogel analogs. This strategy involves the copolymerization of HEMA with 2-aminoethyl methacrylate (AEMA), which displays an amino function usable for the subsequent attachment of peptides of interest [32, 33]. In particular, in recent studies of Kubinova et al., the P(HEMA-co-AEMA) hydrogel was reacted with γ-thiobutyrolactone to introduce thiol groups (SH), which after activation with 2,2′-dithiodipyridine (TPy) immobilized the Ac-CGGASIKVAVS sequence via the SH groups of cysteine (Fig. 1B, top pathway). They noted a significantly higher MSC density and cell growth area on Ac-CGGASIKVAVS-modified P(HEMA-co-AEMA) than that on PHEMA, P(HEMA-co-AEMA), P(HEMA-co-AEMA)-SH and P(HEMA-co-AEMA)-TPy hydrogels. Additionally, the data suggest that the Ac-CGGASIKVAVS-modification enhanced the attachment, proliferation and differentiation of NSCs. Indeed, after two weeks of culture, only NSCs growing on the Ac-CGGASIKVAVS-modified P(HEMA-co-AEMA) hydrogel formed long processes and displayed a neuron-like morphology. In a further study, Kubinova et al. noted in a complete transection model that the implantation of a similar SIKVAV-modified PHEMA hydrogel exhibiting oriented pores, obtained by the use of parallel needle-like ammonium oxalate crystals, allowed axonal fibers to bridge the spinal cord gap [34]. However, the authors pointed out that due to the large size of the Ac-CGGASIKVAVS, it was expected that the peptide did not penetrate into the hydrogel but mostly remained on its surface. In a similar approach, thiol-terminated laminin-derived oligopeptides (CDPGYIGSR and CQAASIKVAV) have been inserted into hydrogels using a sulfosuccinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (sulfo-SMCC) approach. While the sulfo-NHS end group of the sulfo-SMCC reacted with the primary amine groups on the hydrogel, its maleimide end-group reacted with the oligopeptide thiol group (Fig. 1B, bottom pathway). The authors noticed that this peptide-modified hydrogel promoted cell adhesion and neurite outgrowth of primary chick dorsal root ganglia cells in contrast to P(HEMA-co-AEMA) controls [33]. The authors noted the importance of introducing spacers (e.g. additional amino acids) between the biologically active peptide sequence and the polymer chain to improve recognition of receptors (integrins) and thus enhance cellular interactions [35].
Biomolecules other than adhesion peptides were also introduced in the PHEMA in order to optimize hydrogel surface properties, using a similar post-polymerization approach. Růžička et al. have synthesized a serotonin-modified PHEMA (PHEMA-5HT) hydrogel [36], as this neurotransmitter has been shown to support cell attachment and to facilitate neuronal differenciation. In the first step, a porous hydrogel was synthesized by the radical polymerization of HEMA, [2-(methacryloyloxy)ethyl](trimethyl) ammonium chloride (PMADQUAT), 2-aminopropyl methacrylamide hydrochloride (AMPA), and ethylene glycol dimethacrylate (EGDMA) as a cross-linker using azobis(isobutyronitrile) (AIBN) as an initiator and sodium chloride as a porogen. In the second step, after deprotonation, the pendant primary amino groups of the resulting gel were allowed to react with PEG diglycidyl ether, leading to pendant epoxide moieties. The latter were involved in the reaction with the amino group of the serotonin, leading to the PHEMA-5HT. The authors first observed, in vitro, a positive effect of the PHEMA-5HT hydrogel on the growth and differentiation of the seeded cells (i.e. a specific cell line of fetal NSCs derived from fetal human spinal cords, namely SPC-01). Then, after having implanted the SPC-01 cell seeded PHEMA-serotonin hydrogel into a lateral hemisection cavity, they confirmed that the cells survived, proliferated and differentiated into relevant neuronal subtypes. The paracrine effect of those cells protected the remaining tissue and promoted axonal regeneration and angiogenesis. Additionally, the polymer reduced the astrogliosis one month after the injury. However, the absence of functional recovery noticed 3 months after the implantation was explained as a result of enzymatic degradation of serotonin.
PEG-based hydrogels, as attractive candidates for neural tissue repair, have been also the focus of numerous studies regarding peptide functionalization for improving their cell adhesion properties, which are inherently relatively poor. Due to its polyether backbone structure, PEG can only be end-functionalized, in contrast to the above-mentioned PHPMA/PHEMA-based hydrogels in which the radical polymerization process allows introduction of reactive monomers for peptide introduction along the backbone. In this regard, the group of Langer developed an interesting strategy for hydrogel synthesis and peptide incorporation, relying on the Michael addition based reactions between 3-arm thiol-terminated PEG and diacrylate terminated one [37]. The introduction of an oligolysine peptide presenting thiol-containing cysteine end residues (N- and C-terminus) during this process led to successful peptide functionalization of the hydrogel (Fig. 2A), which was shown to promote cell adhesion in a concentration dependent manner. The oligolysine was here chosen as a ‘proof of concept’ model adhesion peptide. In further studies, Shepard et al. similarly reported PEG-based hydrogels functionalized with both RGD and oligolysine peptides [38]. In this case, the oligolysine residues were used as affinity peptides to deliver a gene encoding for the nerve growth factor (NGF), through electrostatic interactions between the cationic lysine residues and the negatively charged plasmid DNA gene. The hydrogel was formed on the same basis of Michael addition, by reaction of the free thiols of cysteine residues of mono-functional or tri-functional peptides with unsaturated vinyl-sulfone moieties present at the chain ends of a 4-arm PEG. The functionalized hydrogel was shown to be efficient at retaining the DNA plasmid and promoted improved neurite outgrowth, as a result of the expression of the inductive NGF protein. More recently, Anseth et al. designed in a very nice study PEG hydrogels functionalized with RGD and YIGSR adhesion peptide sequences, using UV-initiated radical thiol-ene chemistry [39]. The approach relied on the reaction of vinyl group of norbornene moieties present on 4-arm PEG end-chains with (i) thiol functions at both extremities of a peptide (MMP-degradable sequence or oligolysine), for inducing cross-linking, and (ii) thiol functions of the cysteine end residues of CRGD and CYIGSR adhesion peptides (Fig. 2B). The strategy resulted in robust extension of motor axons, from a culture of mouse embryonic stem cell-derived motor neurons (ESMNs). Finally, very recently, an alternative chemical approach based on azide/alkyne click chemistry and microwave-assisted synthesis was reported to prepare agarose-polyacrylic acid (Carbomer)-PEG (ACPEG) based hydrogels functionalized with RGD [40]. This method involved a copper catalyzed azide/alkyne cycloaddition (CuAAC) click reaction between the RGD azide derivative and polyacrylic acid (PAA)- and PEG-modified alkynes. Cross-linking then occurred through microwave-assisted polycondensation reactions of the carboxylic groups of PAA-RGD with the hydroxyl groups of agarose and hydroxyl-end group of PEG-RGD. However, the effect of this hydrogel on cell survival has not yet been studied.
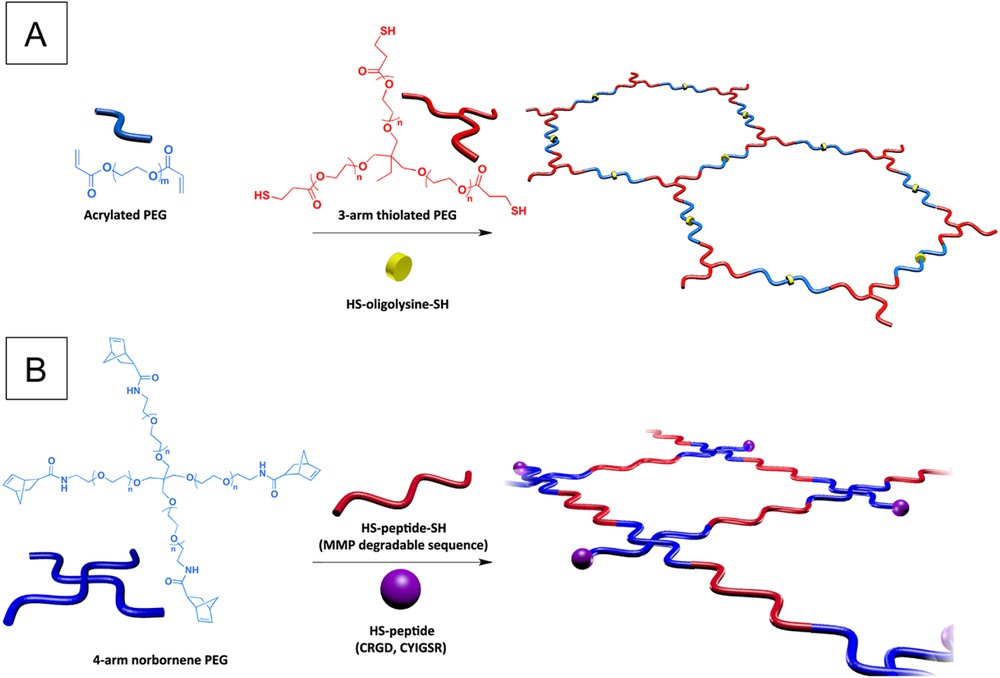
Chemical approaches for PEG-based hydrogel functionalization with cell adhesion peptides; (A) through Michael addition based reactions; (B) through UV-initiated thiol-ene chemistry.
Besides these polymer-based hydrogels, original studies have focused on self-assembled peptide hydrogels. Berns et al. have particularly developed peptide amphiphiles (PAs) based on a hydrophobic polmitoyl moiety attached to a hydrophilic peptide containing an adhesion-promoting sequence [41]. These compounds, namely palmitoyl-VVAAEEEEGIKVAV and palmitoyl-VVAAEERGDS, were obtained through the reaction of palmitic acid with N-terminus of the peptide segment as a last step of the solid phase peptide synthesis (SPPS). Self-assembly of these PAs in water led to hydrogels made of aligned peptide nanofibers over macroscopic length scales. These RGD and IKVAV based hydrogels were found to induce an enhanced neurite growth from neurons (encapsulated in the gel), as compared to the hydrogels made from the scrambled PA analogs (namely palmitoyl-VVAAEEEEGVVIAK and palmitoyl-VVAAEERSDG), unambiguously showing the positive impact of the adhesion peptide sequence. Additionally, the alignment guided the neurite growth along the direction of the nanofibers. Scaffolds encapsulating neural progenitor cells were formed in situ within the spinal cord and resulted in the growth of oriented processes in vivo.
3 Drug delivering hydrogels
Numerous drugs have been identified in the past few years to have a valuable therapeutic potential for SCI. Basically, their actions aim to favor axonal regeneration, cellular survival and to reduce glial scar formation [42]. The direct promotion of neuronal survival, neurite outgrowth and synaptic formation can be induced by various neurotrophic factors such as among others, brain derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), glial cell line-derived neurotrophic factor (GDNF) and nerve growth factor (NGF) [43]. Another strategy is to reduce the effect of neurite outgrowth inhibitors that are generally myelin-associated molecules like myelin-associated glycoprotein (MAG), oligodendrocyte myelin glycoprotein (OMgp) and Nogo-66 [44]. For example, the intrathecal administration of NEP1-40 (i.e., Nogo-66 antagonist peptide) promoted axonal regrowth and functional improvement in rats after a spinal cord hemisection [45]. Enhancing axonal regeneration can also be achieved by limiting the steric hindrance of chondroitin sulfate proteoglycans (CSPGs) through the use of chondroitinase ABC [46, 47]. Recent studies have particularly focused on an anti-glial scarring drug strategy, since it has been shown to be of crucial importance to promote functional recovery after neural injury. Several teams have particularly pointed out the potential of adenosine-based molecules for limiting glial scar formation and promoting axonal regrowth [48–51].
However, the efficiency of these drugs, when delivered in conventional ways (e.g. systemic injection) is generally largely compromised by the rapid bloodstream clearance, the lack of targeting of the injured tissue and the presence of the blood-spinal cord barrier (BSCB). Similarly, drug-loaded nano-carriers (e.g. biodegradable nanoparticles), that have been shown to selectively target the lesion site and to bring substantial improvements [52], still present a limited efficiency that is frequently due to inadequate drug release from the nanoparticles or the difficulty to cross the BSCB [53]. As for local delivery through intrathecal injection, it is not really satisfactory because of rapid clearance due to cerebrospinal fluid flow in the intrathecal space, and the necessity of repeated doses, which poses the problem of recurrent infections. Thus, the use of the hydrogel platform for local and sustained delivery of drugs has appeared as a valuable approach to overcome such limitations and has gained increasing attention in recent research in SCI.
3.1 Controlled release of physically entrapped drugs
The potential of the hydrogel depot strategy for tunable/controlled release of drugs had been particularly highlighted in pioneer studies of Shoichet's group, who designed a hyaluronan/methyl cellulose (HAMC) based hydrogel loaded with a cocktail of drugs (having complementary therapeutic action, e.g. neuroregenerative and neuroprotective) encapsulated or not in biodegradable poly(lactide-co-glycolide) (PLGA) nanoparticles [48, 54]. They showed that the time range of drug release could be tuned so as to be adapted for neuroprotective and neuroregenerative treatments. In particular, neuroprotective molecules, for which fast release is desirable, could be released within 4 days by direct loading in the hydrogel, whereas release of neuroregenerative molecules could be extended over 28 days through encapsulation in PLGA particles loaded in the gel. Interestingly, they also showed that the prejudicial burst release effect classically observed from particles alone was strongly reduced upon particle loading in the gel, allowing a longer and more linear drug release profile.
Similarly, Sellers et al. showed in a recent study that Pluronic F127-based hydrogel encapsulating anti-thrombin agent-loaded PLGA nanoparticles enabled a controlled drug release and a limited initial burst release as compared to what has been observed with hydrogel and nanoparticles alone [55]. These authors chose the use of an anti-thrombin agent (hirudin), as it is long known that it may suppress inflammation and scar formation [56]. They showed that the controlled delivery of hirudin accelerated functional recovery following lateralized demyelination lesion in the mouse spinal cord.
Interesting results have also recently been obtained by Rossi's team on hydrogel-nanoparticle composite systems regarding the ability to finely tune the release of drugs, using polyacrylic acid/agarose based hydrogels and poly(methyl methacrylate) (PMMA) nanoparticles [57]. Indeed, the authors demonstrated the beneficial outcomes of nanoparticle drug encapsulation by confirming the prolonged release and the strongly reduced burst effect of Rhodamine, which is a model drug with similar steric hindrance as anti-inflammatory drugs. The authors have then further studied the in vitro release of different sized nanoparticles (60, 80 and 130 nm) from several entangled hydrogels (30, 60 and 90 nm mesh size). They showed that the diffusion-controlled release of the nanoparticles was highly dependent on nanoparticle size, and to a very less extent on hydrogel mesh size. Indeed, after the intrathecal injection of 2 composites of different sized nanoparticles (60 and 130 nm), a faster nanoparticle release, in agreement with the in vitro result, has been observed for the smaller nanoparticles. These release/diffusion studies, rationalized by mathematical modeling, are of high interest regarding the design of biomaterial library able to satisfy different drug delivery needs. Particularly, the authors have further showed the potential of such nanoparticle-loaded hydrogel for delivering pharmacological compounds in activated microglia/macrophages thus counteracting relevant secondary inflammatory events in SCI [58].
Other original research studies have been focused on diblock copolypeptides (DCHs) as promising hydrogel depots for sustained and controlled delivery of hydrophobic drugs in an injured spinal cord [59]. These block copolypeptides consist of one long hydrophilic and charged block composed of poly(l-lysine) and another short block made of a hydrophobic amino acid sequence (such as l-alanine, l-leucine, l-phenylalanine), and are prepared by sequential ring-opening polymerization of corresponding amino acid N-carboxyanhydride monomers. These peptide-based architectures have been proved to self-assemble at the nano-scale, through a sophisticated mechanism (association of the β-strands and coiling of helices), resulting in hydrogel properties [60]. Interestingly, it was shown that different kinds of hydrophobic compounds can be loaded into DCH and released over prolonged and controllable time frames, and that both the loading capacity and release profiles can be predictably, easily and finely tuned by adjusting the hydrophobic block composition and DCH concentration in water. In addition, in vivo studies revealed that DCH depots could deliver hydrophobic molecules (tamoxifen) that alter gene expression of central nervous system cells in a locally restricted and predicted manner after injection into the forebrain or after a SCI. These non-toxic peptide hydrogels are particularly interesting since they were found to be biodegradable in vivo [61].
3.2 ‘Stimuli-responsive’ controlled release of covalently entrapped drugs
Stimuli-responsive hydrogels are also very promising systems for fine tunable and ‘on-demand’ local drug delivery. In this regard, Shah et al. have developed a remarkable photosensitive system composed of a photo-triggerable drug conjugated silica nanoparticles encapsulated within a PEG-based hydrogel [62]. The rationale behind this strategy is to avoid passive and non-desired diffusion/release of the drug that occurs in the classically reported photo-responsive (e.g., photodegradable) hydrogels. Indeed, this system generally contains non-covalently entrapped drugs that can slightly diffuse in the absence of irradiation, thus leading to poor controllability and poor ratio of drug release between the ‘on’ (light) and ‘off’ (no light) state. The model drug was covalently attached to the nanoparticles through an ortho-nitrobenzyl compound linkage, known to induce a series of chemical rearrangements upon photo-irradiation, which ultimately releases the covalently bound drug. The drug-nanoparticle conjugate was then encapsulated in the hydrogel during its chemical cross-linking through a click reaction between a 4-arm azide end-functionalized PEG and a di-alkyne functionalized linear PEG in water. The potential of this system has been demonstrated for delivery of a model drug to a malignant cancer line, but it is clear that this hybrid scaffold can be easily tuned for other soft tissue engineering/drug delivery applications, such as SCI.
In another stimulus-based strategy, Chu et al. have recently designed a HAMC hydrogel loaded with a PHPMA-bivalirudin based polymer-peptide conjugate. Such a material is able to release bivalirudin (i.e. an anti-thrombin agent) via an enzyme-mediated process [63]. Indeed, the grafted bivalirudin peptide (BM9) was designed to contain an optimized matrix metalloproteinase-9 (MMP9) peptide substrate linker (PRQITAG). The MMP9 was selected as the target protease for triggering bivalirudin release due to its maximal expression at 24 h following SCI. The conjugate preparation consisted of three steps (i) Reversible Addition-Fragmentation Chain Transfer (RAFT) copolymerization of HPMA and N-(3-aminopropyl) methacrylamide hydrochloride (APMA), (ii) modification of the pendant amine groups of the prepared copolymer with a succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC) difunctional agent, leading to the maleimide pendant functions on the copolymer and (iii) coupling of the bivalirudin peptide on the PHPMA copolymer through thiol-maleimide chemistry. This polymer conjugation approach thus allowed an enzyme-mediated release upon MMP9 exposure, and a prolonged release from HAMC hydrogels compared to the free bivalirudin peptide. Localized administration of bivalirudin copolymers in vivo at the site of rat SCI, in a contusion model, decreased cellular proliferation and astrogliosis, suggesting that this bivalirudin copolymer-hydrogel composite system could be a promising therapeutic tool for reducing immediate inflammatory responses and long-term scarring.
4 Degradability
Although the most used synthetic polymer hydrogels (e.g. PHEMA, PHPMA, and PEG) present very interesting features, such as high water content, mechanical stability, soft tissue similarities, and proving support for axonal regeneration, they are non-degradable. Biodegradability is a highly desirable property for any scaffold used in tissue engineering applications, since it is expected to promote tissue regeneration while gradually degrading so that only host tissues remain. In non-degradable hydrogels, axonal regrowth is unfortunately restricted to the existing pores of the biomaterial [3]. Additionally, as these cross-linked hydrogels cannot be cleared from the body, the calcification and prolonged inflammatory response might limit long-term axonal regeneration [64].
Only few past studies have been dedicated to impart biodegradability in these hydrogels. Regarding PEG-based hydrogels, Piantino et al. proposed a versatile approach based on the modification of PEG-end groups with hydrolyzable lactide units terminated by a double bond (acrylated lactide). The latter allowed radical photo-polymerization and cross-linking using an appropriate photo-initiator [65]. Regarding PHEMA-based hydrogels, Atzet et al. reported an approach consisting of polymerizing HEMA in the presence of degradable oligocaprolactone diacrylate as a cross-linker, through an atom transfer radical polymerization (ATRP) process initiated by an oligocaprolactone di-bromide [66]. In the above-mentioned systems, the hydrogel degradation is induced by hydrolytic cleavage of ester containing units (lactide, caprolactone), leading to recover individual polymer chains, which can be cleared from body through renal clearance if present in a sufficiently low molecular weight. Rather than this hydrolytic-based process, interesting alternative strategies relying on enzymatic degradation have been very recently developed. Wang's team has, for example, developed PEG-based copolymers containing polyurethane segments (poly(serinol hexamethylene urethane)) [67]. The latter allowed enzymatic degradation of the hydrogel in the presence of cholesterol esterases. They showed, in a spinal cord contusion model, that transplantation of bone marrow stromal cells (BMSCs) in hydrogel enhanced cell survival and led to superior locomotor and sensory-motor recovery [68]. In another study, Jones et al. reported the preparation of PEG hydrogels containing a collagenase-sensitive peptide, allowing for hydrogel degradation in the ECM microenvironment [69]. Identically as described in the first section, the cross-linked hydrogel was prepared from Michael addition reactions between a 4-arm PEG acrylate and a thiol-terminated peptide as a cross-linker (cysteine at N- and C-terminus) [37]. In a similar chemical approach, based on thiol-ene chemistry, Anseth et al. introduced matrix metalloproteinase (MMP) degradable peptides in their PEG hydrogels, which had a beneficial impact on the axon outgrowth, by allowing the neurons to remodel their extracellular environment [39].
Aliphatic polyesters such as polylactic acid (PLA) and polycaprolactone are interesting biomaterials regarding tissue engineering applications, due to their biodegradability and recognized biocompatibility [70]. However, such biomaterials are too rigid to be implanted into soft neural tissues as they are. Therefore, in a very recent study, we synthesized a PLA-b-PHEMA block copolymer hydrogel in order to benefit from the complementary properties of PHEMA and PLA that are, respectively, softness and degradability [71]. We opted for a block copolymer design, as a physical blending of the two incompatible homopolymers would only result in a non-valid biomaterial displaying a macrophase separation. Our synthesis approach, based on a previous work realized in our laboratory [72, 73], relied on the use of a PLA-SG1 (N-tert-butyl-N-(1-diethylphosphono-2,2-dimethylpropyl)aminoxyl) macroalkoxyamine. Such a molecule is able to initiate polymerization of HEMA (Scheme 1) through nitroxide-mediated polymerization (NMP), which is usually used as a controlled/living radical polymerization technique [74, 75]. The PLA-SG1 was prepared from the intermolecular radical addition of the MAMA-SG1 (BlocBuilder®) alkoxyamine onto acrylate end-capped PLA previously prepared by ring-opening polymerization (ROP).
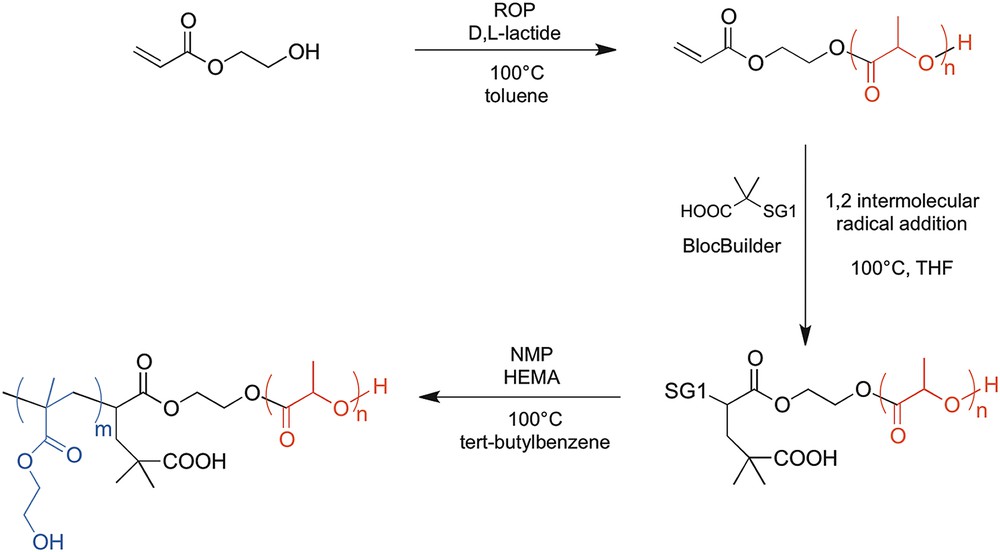
Chemical pathway for preparation of the PLA-b-PHEMA block copolymer through ROP/NMP combination (SG1 = ON(tBu)CH(tBu)PO(OEt)2).
Through an in vitro study, we confirmed that our PLA-b-PHEMA (65/35%w) block copolymer was degradable, as shown by its mass loss over time (Fig. 3), and supported embryonic rat spinal motoneuron survival and neurite outgrowth. Interestingly, the block copolymer could be processed in a porous tridimensional hydrogel, without the need for any cross-linking agent [76], using a versatile and eco-friendly freeze-extraction process, requiring only dimethyl sulfoxide as an organic solvent for block copolymer dissolution, and water for extraction. Once the scaffold is obtained, we implanted it in a hemisected rat spinal cord cavity and observed an axonal regeneration into the hydrogel 8 weeks post-injury (Fig. 4A) and a functional recovery by the use of the Basso, Beattie and Bresnahan (BBB) test, which is a well-known locomotor rating scale (Fig. 4B) [77]. Indeed, although the hydrogel-implanted group did not fully recover, a superior functional improvement has been detected compared to non-treated hemisected rats. Other recent studies on aliphatic polyester-based biomaterials for spinal cord repair consist of increasing polymer flexibility through introduction of trimethylene carbonate (TMC) units along aliphatic polyesters [78]. In a further study, it was demonstated that a poly(trimethylene carbonate-co-ɛ-caprolactone) (P(TMC-CL)) copolymer significantly promote axonal growth [79]. Interestingly, Xing et al. showed the possibility of chemical derivatization of the TMC monomer with bioactive molecules, such as acetylcholine analog. After copolymerization of this TMC modified-monomer with lactide in various proportions, the obtained copolymers promoted neurite outgrowth in a concentration dependent manner [80].
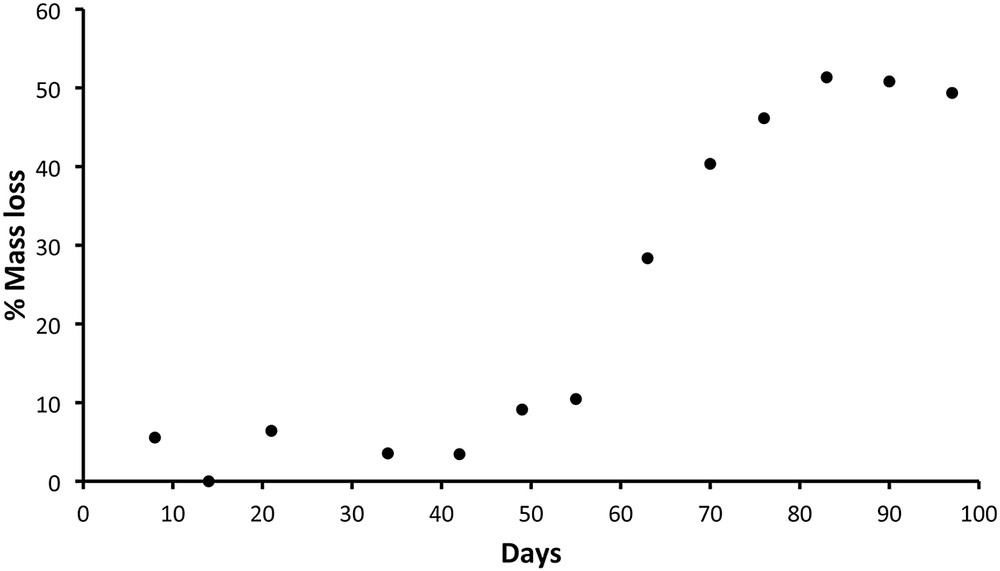
PLA-b-PHEMA block copolymer’s mass loss (expressed as a percentage of its initial mass) as a function of time. While the block copolymer maintained a constant mass over the first 55 days, an increasing mass loss was observed after that period. Indeed, the mass loss reached about 60% after 97 days, consistent with the escape of most part of the PLA degradation residues. Reproduced from Ref. [71] with permission from Elsevier.
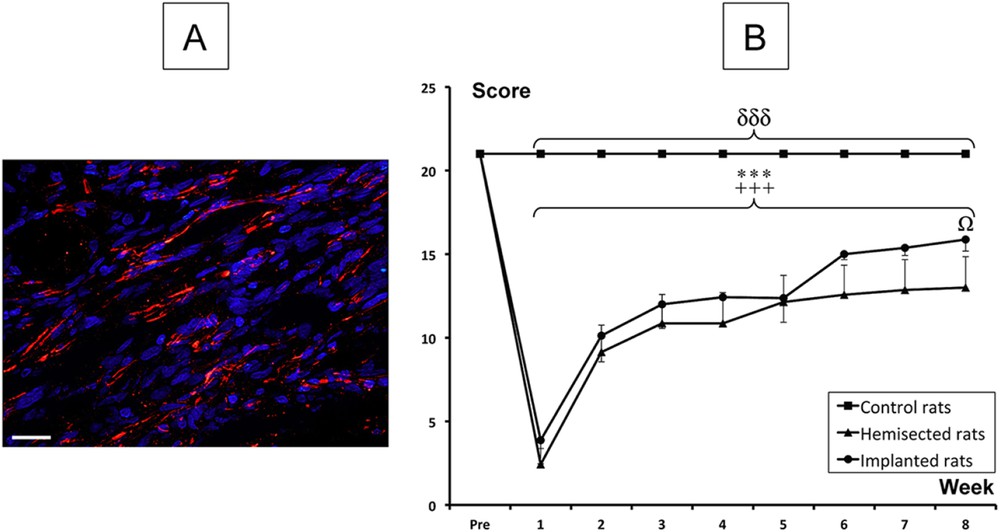
Therapeutic effect induced by the implantation of our PLA-b-PHEMA hydrogel after a spinal cord hemisection. (A) Histological analyses, combining nuclear staining (blue) and axonal marker (red), in the hydrogel region of implanted rats (Scale bar: 20 mm). (B) BBB locomotor rating score. Significant differences in the BBB scores are indicated by * (Hemisected rats pre-injury vs. Hemisected post-injury), + (Implanted rats pre-injury vs. Implanted rats post-injury), δ (Control rats vs. Hemisected and Implanted rats) and Ω (Hemisected rats vs. Implanted rats) (One symbol p < 0.05 and 3 symbols p < 0.001). Adapted from Ref. [71] with permission from Elsevier.
5 Conclusion
In this short review, we exposed the latest chemical strategies that have been developed to potentiate the key properties of synthetic hydrogels intended to be implanted after SCI. By taking profit of the synthetic polymer versatility, several authors improved the hydrogel integration and neurite outgrowth via the coupling of proteins and peptides onto the hydrogel’s surface. Other efforts were devoted to tuning drug release by embedding drug-loaded nanoparticles into the hydrogel, or using relevant stimulus (light, enzyme) responsive hydrogels. Moreover, hydrolyzable and enzyme-sensitive degradable hydrogels were developed so as to avoid chronic complications and to fit with the axon regeneration process.
However, combining all desired characteristics (e.g. biocompatibility, bioadhesion, porosity and degradability) into a single therapeutically efficient biomaterial is complex. For example, it was previously shown that the biomaterial degradation alters its three-dimensional structure that, in turn, affects its capacity to induce axonal regrowth [81]. Therefore, those interdependent characteristics require compromises in the search of the ideal scaffold.
Nevertheless, although clinical trials are not yet conceivable, highly tunable synthetic hydrogels seem to be a perfectly fitted platform for promising combinatory strategies.
Acknowledgements
The authors thank Aix-Marseille Université and the Centre National de la Recherche Scientifique (CNRS) for financial support.