

1 Introduction
Hydrazones are a versatile class of compounds because of their ease of synthesis, their stability and their structural variety, which found numerous applications as valuable organic intermediates [1], molecular components of materials [2] or in acid-degradable polymers for drug delivery [3]. They are also interesting ligands [4] with various applications as sensors [5], in biology [6] or in catalysis [7]. Amongst the large variety of hydrazones, ferrocenyl hydrazones [8] have a particular interest because of the specific properties of the ferrocene moiety (redox properties, planar chirality…). In our ongoing program dedicated to the chemistry of ferrocene derivatives [9], in particular as ligands [10], we present in this paper the synthesis and the full characterization of four new ferrocenyl bishydrazone compounds.
2 Experimental
2.1 General considerations
1,1′-ferrocenedicarboxaldehyde was synthesized by a published procedure [11]. Hydrazines are commercially available and have been used without prior purification. 1D- and 2D-NMR spectra were recorded on a Bruker AV400 spectrometer. 1H and 13C chemicals shifts (δ) are given in ppm (the residual peak of deuterated solvent was used as reference). Peaks are labelled as singlet (s), doublet (d), triplet (t), multiplet (m), and broad (br). The proton and carbon assignments were performed by COSY, HSQC, 1H–13C HMBC experiments. MS spectra were performed by the mass spectrometry service of the Paul-Sabatier University, Toulouse.
2.2 Synthesis of hydrazones
General procedure: in a Schlenk tube, under argon, were added 115 mg (0,47 mmol) of 1,1′-ferrocenedicarboxaldehyde, 625 mg (5,19 mmol) of anhydrous magnesium sulphate MgSO4 and 10 ml of anhydrous dichloromethane. To the red suspension was then added 12 equiv of the specific hydrazine using a syringe. The reaction mixture was then stirred at room temperature overnight. The crude material was purified by flash chromatography on silica gel.
2a: reaction with 430 μL of N,N-dimethylhydrazine to yield 120 mg of 2a as a red solid (yield = 77%). 1H (300 MHz, CDCl3): 7.00 (s, 2H, CH), 4.44 (t, J = 1.8 Hz, 4H, Cp), 4. 14 (t, J = 1.8 Hz, 4H, Cp), 2.81 (s, 12H, CH3). 13C (300 MHz, CDCl3): 133.6 (s, CH), 83.5 (s, quat. Cp), 69.6 (s, subst. Cp), 67.3 (s, subst. Cp), 43.2 (s, CH3). HR MS (ESI+): 327.1266 (100%, 327.1272 for C16H23Fe N4: M + H).
2b: reaction with 940 μL of, N,N-diphenylhydrazine to yield 237 mg of 2b (yield = 87%). 1H (300 MHz, CDCl3): 7.40 (m, 8H, Ph), 7.22–7.16 (m, 12H, Ph), 7.00 (s, 2H, CH), 4.56 (t, J = 1.8 Hz, 4H, Ph), 4.30 (t, J = 1.8 Hz, 4H, Cp). 13C (300 MHz, CDCl3): 144.0 (s, quat. Ph), 135.5 (s, CH), 129.8 (s, Ph), 124.1 (s, Ph), 122.5 (s, Ph), 82.9 (s, quat. Cp), 70.3 (s, subst. Cp), 67.7 (s, subst. Cp). HR MS (ESI): 574.1826 (100%, 574.1821 for C36H30FeN4: M).
2c: reaction with 1 mL of N,N-phenylbenzylhydrazine to yield 286 mg of 2c (yield = 84%). 1H (300 MHz, CDCl3): 7.40–7.30 (m, 8H, Ph), 7.20–7.08 (m, 12H, Ph), 6.96 (s, 2H, CH), 4.98 (s, 4H, CH2), 4. 36 (t, J = 1.8 Hz, 4H, Cp), 4.15 (t, J = 1.8 Hz, 4H, Cp). 13C (300 MHz, CDCl3): 147.7 (s, quat. Ph), 136.2 (s, quat. Ph), 132.0 (s, CH), 129.1 (s, Ph), 128.9 (s, Ph), 127.2 (s, Ph), 126.2 (s, Ph), 120.2 (s, Ph), 114.3 (s, Ph), 83.6 (s, quat. Cp), 70.2 (s, subst. Cp), 67.9 (s, subst. Cp), 49.8 (s, CH2). HR MS (ESI): 603.2198 (100%, 603.2212 for C38H35FeN4: M + 1).
2d: reaction with 626 μL of 1-aminopiperidine to yield 189 mg of 2d (yield = 98%). 1H (300 MHz, CDCl3): 7.37 (s, 2H, CH), 4.52 (t, J = 1.9 Hz, 4H, Cp), 4.23 (t, J = 1.9 Hz, 4H, Cp), 3.02 (m, 8H, CH2), 1.75 (m, 8H, CH2), 1.52 (m, 4H, CH2). 13C (300 MHz, CDCl3): 135.8 (s, CH), 83.2 (s, quat. Cp), 70.0 (s, subst. Cp), 67.6 (s, subst. Cp), 52.7 (s, CH2), 25.3 (s, CH2), 24.2 (s, CH2). HR MS (ESI+): 407.1892 (100%, 407.1898 for C22H31FeN4: M + 1).
2.3 Structural characterization by X-ray diffraction analysis on single crystals
Single crystals were obtained by slow diffusion of hexane into a dichloromethane solution of bis(hydrazones) 2. A single crystal of each compound was mounted under inert perfluoropolyether at the tip of glass fibre and cooled in the cryostream of either a Bruker APEXII CCD diffractometer for 2a, 2c and 2d, or an Agilent Technologies GEMINI EOS diffractometer for 2b.
The structures were solved by direct methods (SIR97 [12]) and refined by least-squares procedures on F2 using SHELXL-2013 [13]. All H atoms attached to a carbon were introduced in calculation in idealised positions and treated as riding models. In compound 2a, there is a rather large residual electron density between the nitrogen atoms of the NMe2 moieties of one of the two independent molecules building the asymmetric unit. This residue has no chemical meaning and might result in some impurities within the crystal used. In compound 2d, one of the Cp ring and the ligand attached to it are disordered over two positions. The disordered model has been refined using the tools available in SHELXL-2013. The drawing of the molecules was realized with the help of ORTEP32 [14]. Crystal data and refinement parameters are shown in Table 1.
Crystal data.
| Identification code | 2a | 2b | 2c | 2d |
| Empirical formula | C16H22FeN4 | C36H30FeN4 | C38H34FeN2 | C22 H30 Fe N4 |
| Formula weight | 326.23 | 574.49 | 602.54 | 406.35 |
| Temperature, K | 180(2) | 180(2) | 180(2) | 180(2) |
| Wavelength, Å | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Triclinic | Tetragonal | Monoclinic | Monoclinic |
| Space group | I41/acd | C2/c | P21/n | |
| a, Å | 6.0720(2) | 21.1674(5) | 18.7678(5) | 5.9149(3) |
| b, Å | 15.7812(5) | 21.1674(5) | 8.5986(2) | 34.905(2) |
| c, Å | 17.1640(5) | 25.6287(11) | 19.0702(5) | 9.6770(6) |
| α,° | 72.617(2). | 90 | 90 | 90.0 |
| β,° | 83.9470(10) | 90 | 108.1710(10) | 94.259(2) |
| γ,° | 87.4030(10) | 90 | 90 | 90.0 |
| Volume, Å3 | 1560.68(8) | 11483.2(7) | 2924.01(13) | 1992.4(2) |
| Z | 4 | 16 | 4 | 4 |
| Density (calc), Mg/m3 | 1.388 | 1.329 | 1.369 | 1.355 |
| Abs. coefficient, mm−1 | 0.965 | 0.557 | 0.551 | 0.771 |
| F(000) | 688 | 4800 | 1264 | 864 |
| Crystal size, mm3 | 0.24 × 0.18 × 0.04 | 0.21 × 0.18 × 0.05 | 0.42 × 0.2 × 0.08 | 0.450 × 0.200 × 0.080 |
| Theta range,° | 1.55 to 28.34 | 3.212 to 29.251° | 2.248 to 27.925 | 3.147 to 26.371 |
| Reflections collected | 48,899 | 30,320 | 23,246 | 29,105 |
| Independent reflections (Rint) | 7765 (0.0342) | 3628 (0.0894) | 3506 (0.0215) | 4061 (0.0284) |
| Completeness, % | 99.8 | 99.8 | 100.0 | 99.2 |
| Absorption correction | Multi-scan | Multi-scan | Multi-scan | Multi-scan |
| Max./min. transmission | 0.7453/0.6634 | 1.0/0.633 | 0.7456/0.6830 | 0.7456/0.6871 |
| Refinement method | F2 | F2 | F2 | F2 |
| Data/restraints/parameters | 7765/0/387 | 3628/0/186 | 3506/0/195 | 4061/38/280 |
| Goodness-of-fit on F2 | 1.065 | 1.017 | 1.139 | 1.090 |
| R1, wR2 [I > 2σ(I)] | 0.0530, 0.1274 | 0.0516, 0.0889 | 0.0295, 0.0813 | 0.0372, 0.0838 |
| R1, wR2 (all data) | 0.0682, 0.1360 | 0.1008, 0.1049 | 0.0350, 0.0925 | 0.0437, 0.0878 |
| Residual density, e·Å−3 | 2.958/−0.684 | 0.424/−0.458 | 0.442/−0.299 | 0.372/−0.364 |
Crystallographic data (excluding structure factors) have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication No. CCDC 1041697-1041700. Copies of the data can be obtained free of charge on application to the Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [fax: (+44) 1223 336 033; e mail: deposit@ccdc.cam.ac.uk].
3 Results and discussion
3.1 Synthesis and NMR characterization
A series of ferrocenyl bishydrazones were efficiently synthesized in mild conditions. Good to excellent yields (77–98%) were obtained by reaction of 1,1′-ferrocenedicarboxaldehyde11 and an excess of 1,1-disubstituted hydrazine in the presence of magnesium sulphate working as a Lewis acid and a water trap [15]. The isolated yields are from good to excellent (77–98%) (Scheme 1).

Synthesis of ferrocenyl bis(hydrazones) 2a–2d.
The four new bis(hydrazones) 2a–2d have been fully characterized by 1H, 1H NMR, 2D-1H COSY, 1H–13C HSQC, 1H–13C HMBC experiments. The NMR data clearly show that both aldehyde functions reacted with hydrazine. The four molecules are highly symmetrical. In 1H NMR, only two signals for the ferrocene part are present, showing that both Cp rings are equivalent and that the two hydrogen atoms in α or β positions relative to the CN bond are equivalent. In 13C NMR, only a set of three signals for the ferrocene moiety is observed: ipso carbons (C1 and C1′), α and β carbons. Similarly, only one set of signals for both hydrazone groups is observed in 1H (H11 and signals from R1 and R2) and 13C NMR (C11 and signals from R1 and R2) showing that both hydrazones have the same geometry (Z or E). The observed diastereoisomers have been identified in the solid state by X-ray diffraction analysis as the ones in a trans position relative to the ferrocene part (see below, part 3.2) (Scheme 2).
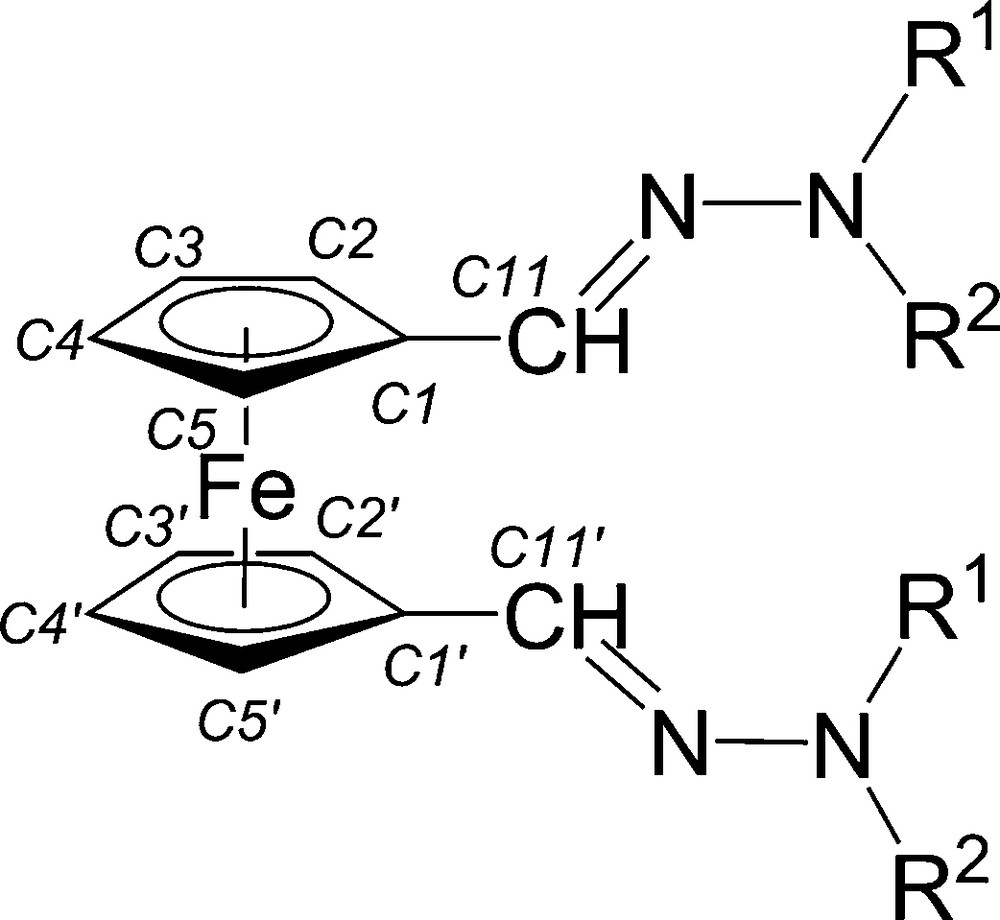
Numbering scheme for ferrocenyl bis(hydrazones) 2a–2d.
3.2 Structural characterization by X-ray diffraction analysis on single crystals
Single crystals have been obtained by slow diffusion of hexane into dichloromethane solutions of the four ferrocenyl bis(hydrazones) 2a–2d and studied by X-ray diffraction analysis. In all four compounds, the ferrocene is substituted in 1,1′ positions by the hydrazone moiety. The arrangement of the two organic chains with respect to each other is greatly dependent on the terminal nitrogen substituents, as indicated by the torsion angles involving the first carbon atoms of the chain and the centroids of the Cp rings (Table 2).
Torsion angles (°) showing the orientation of the hydrazone moieties with respect to the Cp rings.
| Torsion angle (°) | 2a | 2b | 2c | 2d |
| C—Ct1—Ct2—C | 2.65(9) 9.4(1) | 52.76(8) | 140.30(3) | 3.69(8) 13.8(1) |
The most pronounced torsion angles correspond to the bulkier substituents on the terminal nitrogen atom.
The asymmetric unit of compound 2a contains 2 molecules A (Fig. 1) and B. They differ only by the relative conformation of the hydrazone chains in 1′ position on the ferrocene. As shown by molecular fitting [16], the hydrazone chains display opposite conformation in molecules A and B (see Fig. 2). For both molecules, the N-substituents of the CN bond are in trans position relative to the ferrocene part, which allows us to identify the structure of the single isomer observed by NMR in solution.
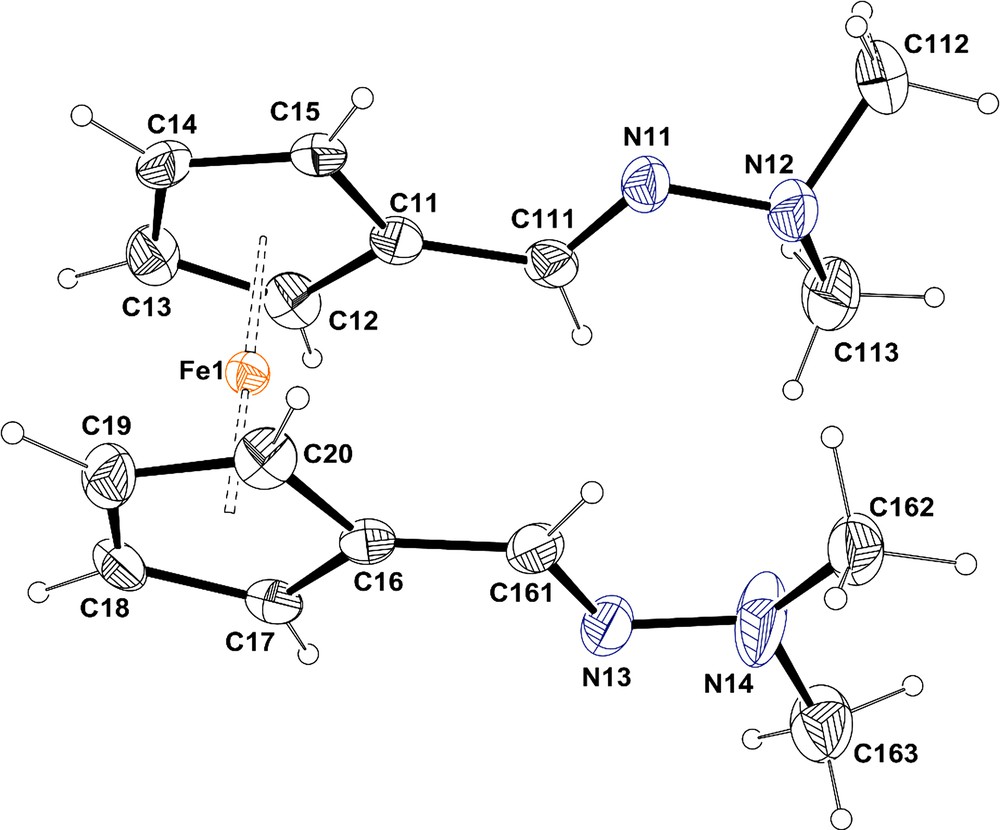
(Colour online.) ORTEP representation of molecule A for compound 2a with the atom labelling scheme. Ellipsoids are drawn at the 50% level. H atoms are represented as small spheres of arbitrary radii.
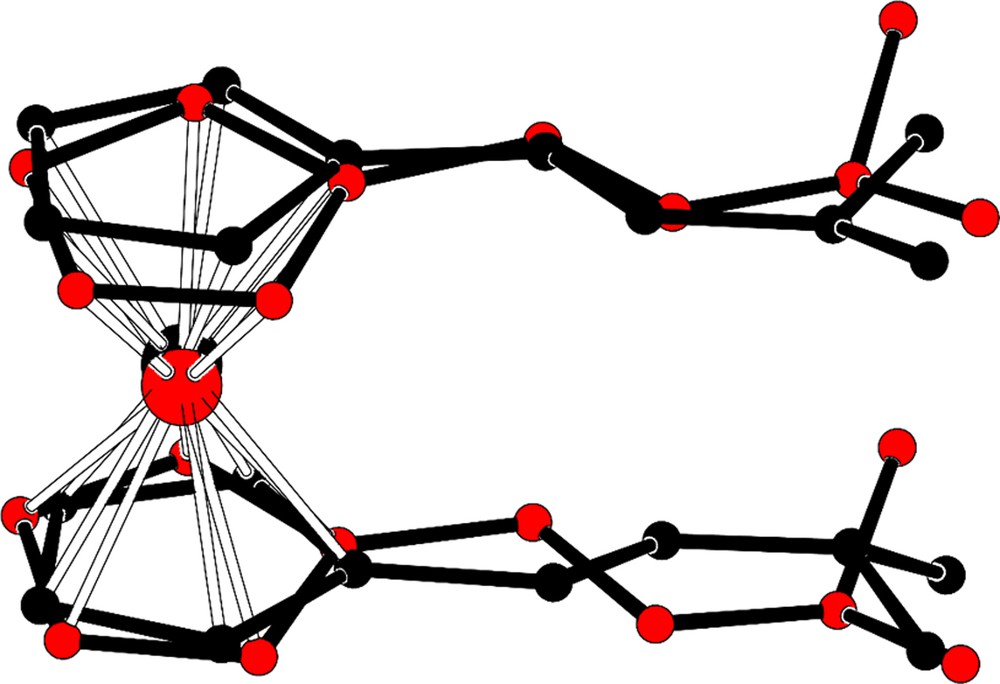
(Colour online.) Molecular fitting of molecules A (black) and B (red) for compound 2a showing the different conformation of the hydrazine moieties.
Indeed, in molecule A, the two hydrazine chains are parallel but crossed, whereas in molecule B both chains are perfectly superimposed.
In both molecules, dimethyl hydrazone chains are in a trans position with respect to the ferrocene group. In molecule A, both Cp rings are slightly staggered with a twist angle of 7.3(2)°, while in molecule B, the two rings are eclipsed with a twist angle of 1.41(2)°. In both molecules, the Cp rings are slightly folded with a dihedral angle of 2.0 (2)° and 2.4 (2)°, respectively, for A and B.
In compound 2b, the iron atom is located on a twofold axis and so the asymmetric unit contains half of the whole molecule (C5H4)2Fe{CN2(C6H5)2}2 (Fig. 3).

ORTEP representation of compounds 2b with the atom labelling scheme. Ellipsoids are drawn at the 50% level. H atoms are represented as small spheres of arbitrary radii [symmetry code: (i) 3/4 + y,–3/4 + x,1/4–z].
Again, only the diastereoisomer with the N-substituents of the CN bond in trans position relative to the ferrocene part was observed.
In addition, owing to the bulky diphenyl substituent, the two Cp rings have rotated with respect to the roughly overlapped 1,1′ position in 2a. The C11–Ct1–Cti–C11i torsion angle is 58.0(1)° (symmetry code (i): ¾ + y,–3/4 + x, ¼–z) (Table 3). Considering only the two Cp rings without the substituents, there are slightly staggered with a twist angle of 15.2°. There is a weak π–π interaction between the C(211)–C(216) phenyl ring and its symmetry related (¾ + y,–¾ + x, ¼–z) with a centroid-to-centroid distance of 3.89 Å and a centroid-to-plane distance of 3.62 Å resulting in a slippage of 1.31 Å. Considering only the Cp rings, they are slightly staggered, with a twist angle of 15.6(2)°.
Selected distances (Å) and angles (°) within the hydrazone moieties.
| 2a | 2b | 2c | 2d | |
| C1–C2 | A 1.456(4), 1.449(4) | 1.449(3) | 1.457(2) | 1.458 (3), 1.489(5) |
| B 1.446(4), 1.478(4) | 1.581(7) | |||
| C2–N1 | A 1.285(4), 1.287(4) | 1.285(3) | 1.281(2) | 1.272(3), 1.274(6) |
| B 1.278(4), 1.270(4) | 1.268(6) | |||
| N1–N2 | A 1.376(4), 1.367(4) | 1.375(2) | 1.3742(15) | 1.386(2), 1.385(7) |
| B 1.375(4), 1.381(4) | 1.393(7) | |||
| N2–C3 | A 1.446(5), 1.438(5) | 1.405(3) | 1.456(2) | 1.440(3), 1.465(7) |
| B 1.449(5), 1.443(4) | 1.459(8) | |||
| N2–C4 | A 1.455(5), 1.399(5) | 1.435(3) | 1.401(2) | 1.450(3), 1.462(8) |
| B 1.440(5), 1.440(5) | 1.268(6) | |||
| C1–C2–N1 | A 120.0(3), 120.4(3) | 119.0(2) | 119.86(13) | 120.1(2), 112.5(5) |
| B 120.6(3), 120.5(3) | 110.0(5) | |||
| C2–N1–N2 | A 119.9(3), 119.9(3) | 119.2(2) | 120.58(12) | 120.5(2), 112.5(2) |
| B 119.6(3), 118.1(3) | 119.8(6) | |||
| N1–N2–C3 | A 111.2(3), 120.6(3) | 116.10(18) | 122.84(11) | 119.5(2), 116.9(5) |
| B 119.3(3), 110.5(3) | 119.8(6) | |||
| N1–N2–C4 | A 119.1(3), 114.8(3) | 121.23(18) | 115.69(11) | 110.2(2), 109.5(6) |
| B 110.6(3), 120.7(3) | 108.4(5) |
In compound 2c, the iron atom is located on a twofold axis and there is only half of the molecule within the asymmetric unit (C5H4)2Fe{CN2CH2(C6H5)2}2 (Fig. 4).

ORTEP representation of compounds 2c with the atom labelling scheme. Ellipsoids are drawn at the 50% level. H atoms are represented as small sphere of arbitrary radii. [symmetry code: (i) 1–x, y, ½–z].
Only the diastereoisomer with trans relationships between the N-substituents of the CN bond and the ferrocene part is present in the crystal.
Owing to the more bulky benzyl and phenyl groups substituent, the two hydrazones are pointing in opposite directions, as indicated by the large torsion angle of 140.30(3)° (Table 3). However, the Cp rings are eclipsed, with a twist angle of 1.6(2)°. The Cp rings are slightly folded with a dihedral angle of 2.98(1)°.
In compound 2d (Fig. 5), only the diastereoisomer with the N-substituents of the CN bond in trans position relative to the ferrocene part was also observed. One of the hydrazone CN fragments is statistically disordered over two positions (Scheme 3). As shown in the scheme, such disorder implies a disorder within the Cp and the piperidine rings. The disorder within the Cp has been ignored, only the disorder of the piperidine has been modelled using the tools available in SHELXL-2013. This disorder might result from a co-crystallization of the two forms: one with a nearly parallel conformation of the two hydrazine CCNN, and the second one with a crossing conformation of the two chains as observed in compound 2a.
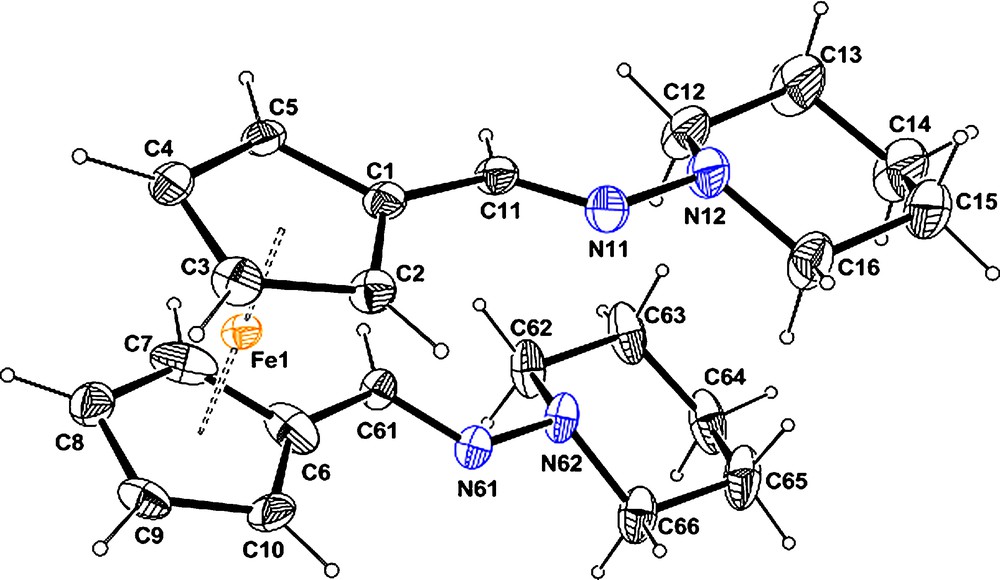
(Colour online.) ORTEP representation of compounds 2d with the atom labelling scheme. Ellipsoids are drawn at the 50% level. H atoms are represented as small sphere of arbitrary radii. Only one component of the disordered moiety is displayed for the sake of clarity.
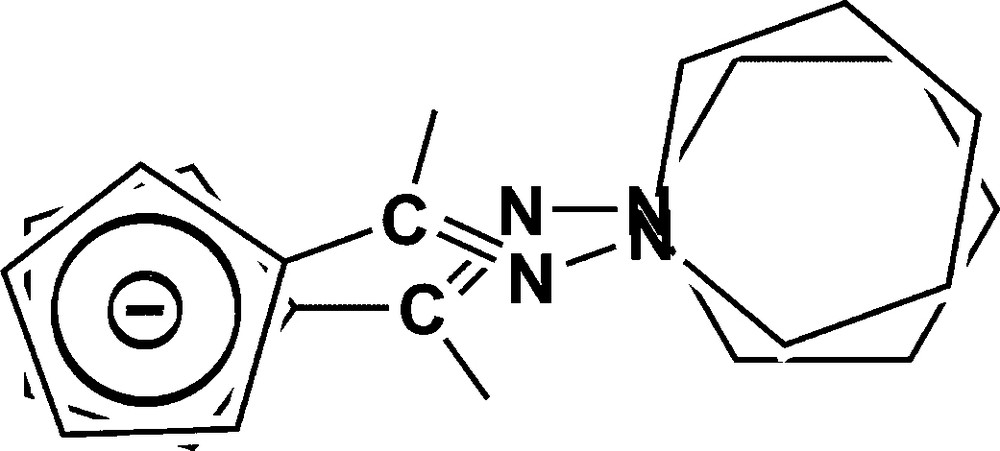
Possible disordered arrangement of the hydrazine in compound 2d.
Owing to the less bulky piperidine group, the two hydrazine are pointing in the same direction, with a torsion angle (Table 3) of 3.69(8)° or 13.8(1)°. The two Cp rings are roughly eclipsed with a twist angle of 4.3(3)°. Owing to the disorder, the two hydrazone chains are either nearly parallel or crossed. The Cp rings are roughly parallel within experimental errors, 1.8(9)°.
Bond distances and bond angles within the chain are identical within the limits of experimental error as shown in Table 3. The values of the distances CN and NN indicate that the double bond is localized between the CN bonds. However, the distance NN, relatively short for a single bond, suggests a partial delocalization along the chains. The slightly different values observed for 2d are certainly related to the influence of the disorder.
4 Conclusion
Four new ferrocenyl bishydrazones have been synthesized by a straightforward method and have been fully characterized by NMR, MS and X-ray diffraction analysis. These new compounds are potentially ligands, and further studies of their coordination chemistry are now in progress in our laboratories.
Acknowledgements
We thank the University Constantine-1 (Algeria) for a grant to NMT. Additional support from the CNRS (France) is also gratefully acknowledged.