

1 Introduction
A vast number of structurally diverse quinones occur naturally; some of them play a major role in the redox electron-transport chains of living systems [1]. In addition, many drugs, such as doxorubicin, daunorubicin and mitomycin C in cancer chemotherapy contain quinones [2], whereas various other quinones have been used in industry [3]. Sayre and co-workers have investigated the behavior of quinone derivatives in the presence of benzylamine as a model for copper amine oxidases [4]. For example, the copper amine oxidases catalyze the oxidative deamination of primary amines to aldehydes at the expense of reduction of O2 to H2O2 [5]. Similarly, quinone-mediated transamination of amines has precedent in the synthetically useful deamination of branched primary amines by 3,5-di-tert-butyl-1,2-benzoquinone [6] and in the deamination of benzylamine by pyrroloquinolinequinone. Sayre, Klinman and co-workers also showed that catalytic deamination of benzylamine and other amines proceeds with the addition of benzylamine to a carbonyl group of TPQ. This process forms a Schiff base, which hydrolyzes to release aldehyde and generates the reduced aminoresorcinol form of the cofactor. The Schiff base is subsequently reoxidized to the starting quinone with the release of NH3 [7]. Amino derivatives of quinones are important building blocks in the synthesis of a variety of natural products, medicinal compounds [8] with antitumor and antimalarial activities [9]. Many of the alkylamino derivatives are involved in enzyme inhibition and DNA cross-linking [10].
The importance of interaction of amines, especially benzylamine derivatives, with quinones has motivated us to study the electrochemical oxidation of hydroquinone in the presence of various types of benzylamines. Aminoquinones are formed in the reactions of different amines with quinones [11]. For example 1,4-benzoquinone reacts with primary amines to give 2,5-diamino-1,4-benzoquinones. A similar reaction of 1,4-naphthoquinone with primary amines results in the formation of 2-amino-1,4-naphthoquinones [11d]. We previously studied the electrochemical oxidation of catechol derivatives in the presence of dibenzylamine [12]. It was shown that the electrochemically generated 1,2-benzoquinones participates in Michael type addition reactions with dibenzylamine, and converts to the amino or diamino substituted 1,2-benzoquinone derivatives. We also recently reported that electrochemical oxidation of 3,5-di-tert-butylcatechol in the presence of benzylamines produces 2-arylbenzoxazoles under green conditions [13]. In this work we describe a new route for the reaction of benzylamine derivatives with electrochemically derived p-benzoquinone.
2 Results and discussion
2.1 Electrochemical study of HQ in the presence of primary amines
The oxidation of hydroquinone (HQ) is pH dependent [14]. Similarly, the concentration of deprotonated benzylamine(s) (BA) depends on the pH [15]. Therefore, studying the effect of pH on the reaction of electrogenerated p-benzoquinone with benzylamines is essential to identify the optimum pH value. Fig. 1a shows the cyclic voltammogram of HQ at pH = 10.0. It is worth mentioning that the pka1 of HQ is 9.85 [16], therefore the HQ monoanion (HQ−) predominates at pH = 10. As can be seen, it shows one anodic peak (A1) and its cathodic peak (C1), which corresponds to the transformation of HQ− to p-benzoquinone and vice versa [17]. The oxidation of HQ− in the presence of BA as a nucleophile was also studied at pH = 10.0. Fig. 1b shows the cyclic voltammogram obtained for a solution of HQ− in the presence of BA. The comparison of cathodic to anodic peak current ratio (IpC1/IpA1) in the absence and presence of BA shows a decrease of the IpC1/IpA1 for the latter (Fig. 1b, pH = 10.0). The existence of a subsequent chemical reaction between p-benzoquinone and BA is supported by the following evidence: (a) The decrease in the IpC1/IpA1 ratio indicates that p-benzoquinone is removed by a chemical reaction with BA. (b) The dependency of IpC1/IpA1 on the potential sweep rate shows that with an increasing sweep rate, the peak current ratio (IpC1/IpA1) increases. It shows a departure from the intermediate region and arrival at the diffusion region [18]. It is worth mentioning that the same electrochemical results were obtained when methoxybenzylamine (MBA) (a benzylamine with an electron donating group) and chlorobenzylamine (CBA) (a benzylamine with an electron withdrawing group) were used.
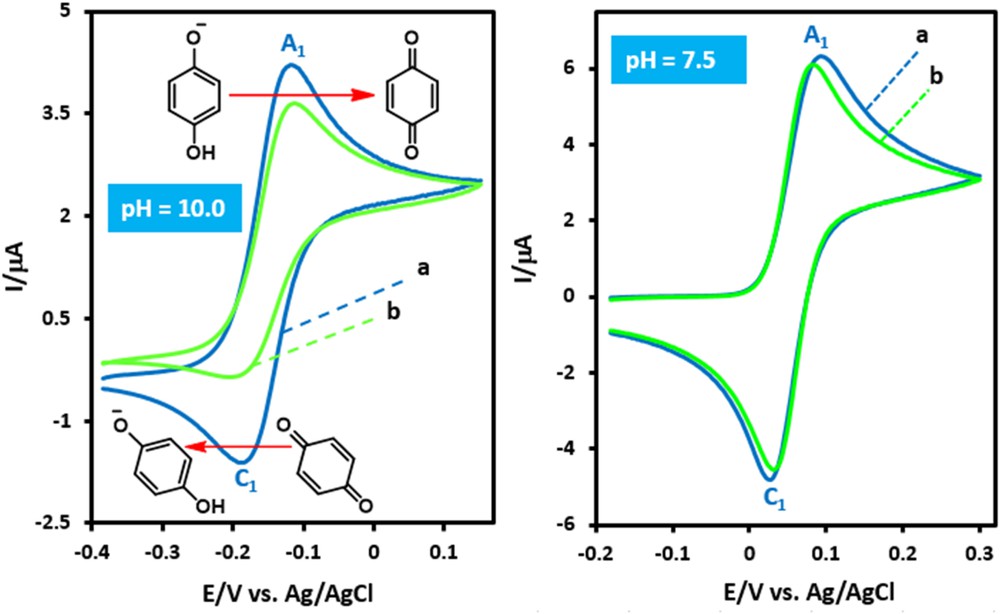
Cyclic voltammograms of hydroquinone (1.0 mM) (a) in the absence, (b) in the presence of benzylamine (4.0 mM), at pH values 10.0 and 7.5, at a scan rate of 10 mV s−1, on a glassy carbon electrode. Temperature = 25 ± 1 °C.
It has been shown that in basic solutions the electrochemically generated quinones are unstable due to side reactions, such as hydroxylation [19] and/or dimerization reactions [20]. The rates of these reactions are pH dependent processes and enhanced by increasing pH [21]. Our result shows that the interference of these reactions is important especially at pH values ≥12. At these pHs, HQ− shows an irreversible anodic process (Fig. 2).

Cyclic voltammogram of hydroquinone (3.0 mM) at pH = 12.0, at a scan rate of 10 mV s−1, on a glassy carbon electrode. Temperature = 25 ± 1 °C.
Fig. 1 also shows the cyclic voltammograms of HQ in the presence and absence of BA at pH 7.5. There is little difference between the cyclic voltammograms of HQ in the absence (curve a) and presence (curve b) of BA. This is attributed to partial protonation of BA and its inactivation toward the Michael type addition reaction with p-benzoquinone. Therefore synthesis at alkaline pH has been avoided and the lower reaction rate was preferred at the lower pH value (pH = 7.5), rather than impure products at higher pH values.
The electrochemical results in conjunction with spectroscopic characterizations (IR, 1H NMR, 13C NMR and mass spectrometry) of the purified samples (see the Section 4) were used to propose a mechanism for the electrochemical oxidation of hydroquinone (HQ) in the presence of benzylamine (BA) (Scheme 1). Interestingly, the electrospray ionization (ESI) mass spectroscopic data of the final product of the HQ/BA system shows 408 Da, indicating a higher mass value for a single nucleophilic attachment on the HQ backbone. Similar trends were observed for HQ/MBA and HQ/CBA systems. The NMR spectroscopic data also confirmed that products with more complicated structural features are Michael type addition reaction products.
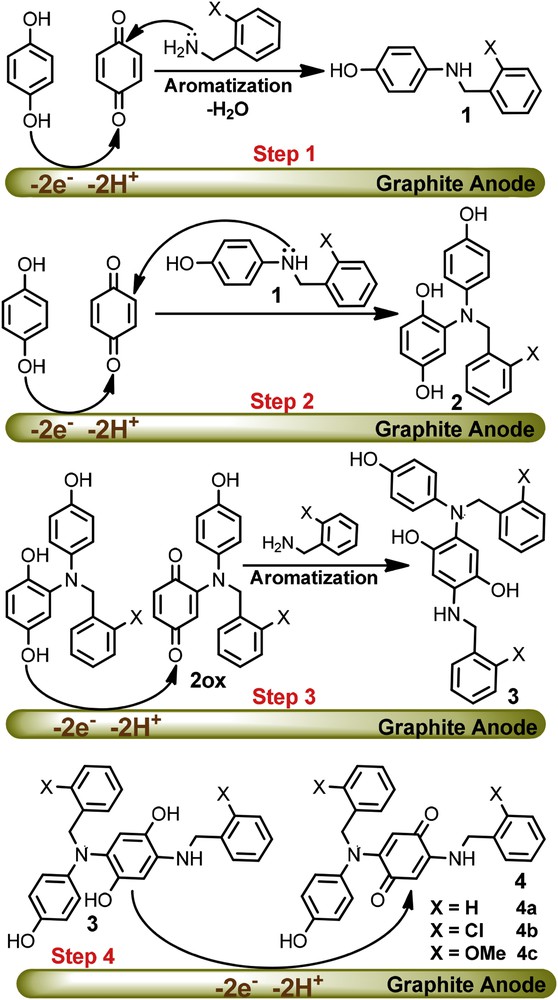
Proposed mechanism for the electrochemical oxidation of hydroquinone in the presence of benzylamine derivatives.
The first step of the proposed mechanism is undoubtedly the electrochemical generation of p-benzoquinone. Once formed, this would be followed by the addition of the benzylic amine to afford a secondary amine, structure 1. In the second step, compound 1 can serve as a nucleophile in a reaction with another p-benzoquinone to form hydroquinone 2. The oxidation of compound 2 is easier than the oxidation of HQ due to the presence of the electron-donating amine group. Therefore, the oxidation of 2 occurs during the electrolysis at the potential of 0.10 V vs. Ag/AgCl (EpA of HQ in pH = 7.5). Consequently in step 3, electrogenerated 2ox would serve as an acceptor in a reaction with BA to form the asymmetric di-substituted hydroquinone 3. The oxidation of 3 is also easier than the oxidation of HQ, therefore it was converted to di-substituted p-benzoquinone 4 in the final step. The 1H NMR spectra revealed, the presence of singlet peaks at δ = 5.27, 5.21 and 5.27 ppm in 1H NMR spectra of products 4a–4c, respectively. These protons are quinonic protons, which support the presence of the substituent groups in the para position of the quinone ring.
2.2 Electrochemical study of HQ in the presence of N-methylbenzylamine
Fig. 3 shows the cyclic voltammograms of HQ in the presence and absence of N-methylbenzylamine (NBA) at pHs 2.0 and 10.0. There is no significant difference between the cyclic voltammograms of HQ in the absence (Fig. 3a) and presence (Fig. 3b) of NBA at pH = 2.0. As discussed, this is related to the protonation of NBA and its inactivation toward the Michael type addition reaction with p-benzoquinone. The rate of the coupling reaction between p-benzoquinone and NBA is pH dependent and enhanced by increasing pH [21]. The cyclic voltammogram of HQ at pH = 10.0 in the presence of NBA shows that the cathodic peak C1 disappears and the anodic peak A1 increases. These observations indicate that a chemical reaction follows the electron transfer process: p-benzoquinone is consumed in a reaction with NBA. However, pH = 7.5 was chosen for bulk electrolysis for minimization of the side reactions such as hydroxylation [19] and/or dimerization [20]. It is worth comparing the reactivity of BA with NBA at pH = 10.0, where the electron donating property of the methyl group in the NBA leads to higher reactivity rather than the BA. According to the electrochemical and spectroscopic data of the product, the proposed mechanism for the oxidation of HQ in the presence of NBA is shown in Scheme 2.

Cyclic voltammograms of hydroquinone (1.0 mM) in the presence of N-methylbenzylamine (4.0 mM) at pHs = 2.0 and 10.0 at a scan rate of 10 mV s−1, on a glassy carbon electrode. Temperature = 25 ± 1 °C.
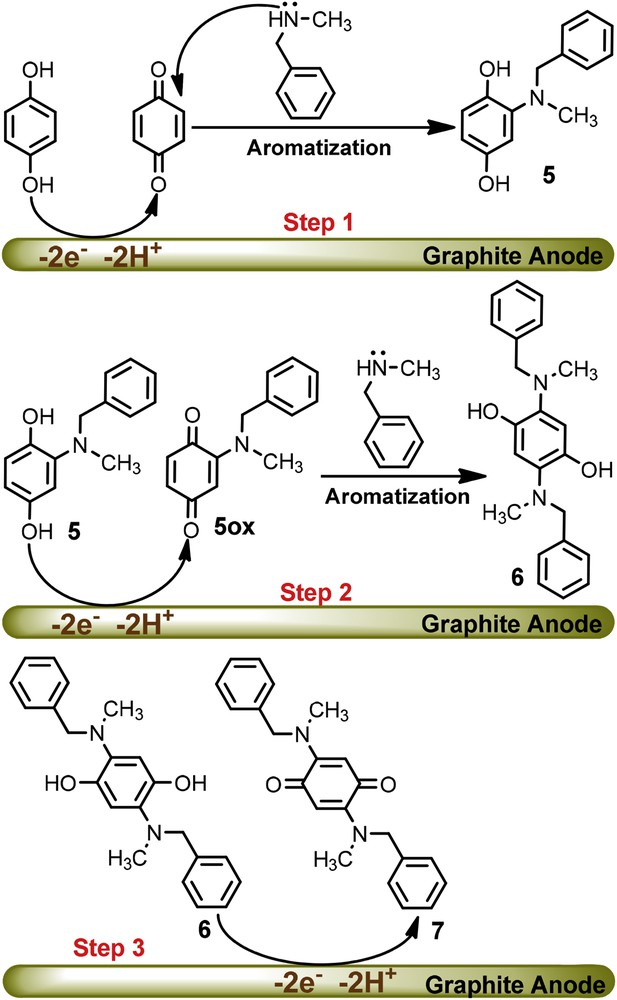
Proposed mechanism for the oxidation of HQ in the presence of NBA.
According to the proposed mechanism, in the first step, the electrochemical generation of p-benzoquinone is followed by a Michael type addition reaction of NBA producing hydroquinone 5. Since, the oxidation of 5 is easier than the oxidation of HQ, its oxidation occurs during the electrolysis at the potential of 0.10 V vs. Ag/AgCl. In the second step, electrogenerated 5ox would serve as an acceptor in a reaction with NBA to form the di-substituted hydroquinone 6. Finally, the oxidation of 6 at the surface of the electrode produces di-substituted p-benzoquinone 7 as a final product. The presence of a singlet peak at δ = 5.42 ppm with the integration 2H (compound 7), which was assigned to quinonic protons, confirms the symmetrical structure of 7 and the presence of the NBA groups in the para position of the quinone ring. The symmetrical structure was further confirmed by considering the number of peaks in the 13C NMR spectrum.
3 Conclusions
We have reported an efficient one-pot method for the synthesis of some new symmetric and asymmetric p-benzoquinone derivatives. This work has several advantages over traditional methods. (i) Neither organic/inorganic oxidizing agents nor catalysts are necessary and the reaction can be performed under mild and green conditions. (ii) It is convenient to carry out and can be done at room temperature and atmospheric pressure. (iii) The atom economy of the reactions is high (94.5–98.3%) showing the incorporation of all reagents into the final products (this also contributes to reduction of waste). This also allowed for the design of a simple separation and purification procedure of both 4a–4c and 7 compounds. The extension of this electrochemical system for the preparation of other organic compounds is under way in our laboratory. Finally, although the experiments were conducted on a relatively small scale, there is little difficulty in producing larger quantities either by using larger cells or by running several cells in series.
4 Experimental
4.1 Apparatus and reagents
Cyclic voltammetry and electrolysis were performed using an Autolab model PGSTAT20 potentiostat/galvanostat. The working electrode was a glassy carbon disk (1.8 diameter), and Pt wire was used as a counter electrode. The working electrode potentials were measured versus Ag/AgCl (3M KCl) (all electrodes from Metrohm). An assembly of four normal soft carbon rods (6 mm diameter and 4 cm length) was used as the working electrodes in macro-scale electrolysis and controlled-potential coulometry. The carbon rods were placed at the edges of a square with a distance of 3 cm, and a large stainless steel cylinder (25 cm2 area) was used as the counter electrode. The glassy carbon electrode was polished using alumina slurry (from Iran Alumina Co.). The final product(s) was mixed with dried KBr powder and a pallet was made to record the infrared (IR) spectra. A Perkin Elmer 1760 spectrometer at a resolution of 2 cm−1 was used. Nuclear Magnetic Resonance (NMR) spectroscopy (1H and 13C) was conducted on a Varian INOVA 400 MHz spectrometer. Hydroquinone (HQ), benzylamine (BA), 2-chlorobenzylamine (2ClBA), 2-methoxybenzylamine (2MBA), and N-methylbenzylamine (NBA) were reagent-grade material from Aldrich and used as received. All other chemicals were purchased from E. Merck. More details are described in a previous paper [22].
4.2 General electrochemical procedure
Aqueous phosphate buffer solution (80 mL, c = 0.2 M, pH = 7.5) containing 0.25 mmol of hydroquinone and 0.25 mmol of benzylamine (2-cholorbenzylamine, 2-methoxybenzylamine or N-methylbenzylamine) was subjected to electrolysis at 0.10 V versus Ag/AgCl, in a divided cell. The electrolysis was terminated when the decay of current became more than 95% of the initial value. The process was interrupted during the electrolysis and the carbon anode was washed in acetone in order to reactivate it. At the end of the electrolysis, the precipitated solid was collected by filtration and was washed several times with water. After drying, the final products were further crystallized in acetone.
4.3 2-(Benzyl(4-hydroxyphenyl)amino)-5-(benzylamino)cyclohexa-2,5-diene-1,4-dione (C26H22N2O3) (4a)
Brown crystals, (67% yield); mp 252–253 °C (dec.); 1H NMR (400 MHz, acetone-d6) δ 1.29 (s, 1H), 4.50 (d, J = 2.8 Hz, 1H), 4.56 (t, J = 2.0 and 4.0 Hz, 2H), 5.27 (s, 1H), 5.99 (s, 2H), 6.92 (dd, J = 9.2, 2.4 Hz, 1H), 7.26 (m, 6H), 7.42 (m, 7H), and 7.57 (d, J = 2.3 Hz, 1H) ppm; 13C NMR (125 MHz, DMSO-d6) δ 45.4, 47.1, 93.1, 96.9, 104.7, 113.4, 115.8, 124.5, 126.6, 127.1, 128.37, 128.41, 132.2, 136.0, 136.9, 137.2, 137.4, 149.7, 155.4, 176.9, and 178.2 ppm; IR (KBr): 3367, 3277, 2924, 2853, 1736, 1645, 1582, 1471, 1455, 1357, 1262, 1219, 1091, 933, 810, 793, 696, and 464 cm−1; MS (EI): m/z (Relative intensity %): 410 [M] (5), 409 [M−H] (18), 408 [M-2H] (59), 318 (79), 227 (11), 213 (18), and 91 (100).
4.4 2-((2-Chlorobenzyl)(4-hydroxyphenyl)amino)-5-((2-chlorobenzyl) amino)cyclohexa-2,5-diene-1,4-dione (C26H20 Cl2N2O3) (4b)
Brown crystals, (62% yield); mp 190–191 °C (dec.); 1H NMR (400 MHz, acetone-d6) δ 1.29 (s, 1H), 4.64 (d, J = 6.4 Hz, 2H), 5.21 (s, 1H), 6.06 (s, 2H), 6.47 (d, J = 8.3 Hz, 1H), 6.94 (dd, J = 8.8, 2.9 Hz, 2H), 7.14 (t, J = 7.6 Hz, 1H), 7.26 (t, J = 7.8 Hz, 2H), 7.33 (m, 4H), 7.48 (m, 4H), and 7.62 (d, J = 2.4 Hz, 1H) ppm; 13C NMR (125 MHz, DMSO-d6) δ 45.3, 47.0, 98.9, 106.9, 114.2, 117.5, 126.4, 127.9, 128.7, 130.1, 130.5, 130.8, 130.9, 134.4, 135.7, 136.1, 150.9, 157.0, 178.6, and 180.2 ppm; IR (KBr): 3330, 1694, 1656, 1595, 1503, 1478, 1443, 1353, 1235, 1196, 1097, 1049, 951, 793, 751, and 457 cm−1; MS (EI): m/z (Relative intensity %): 478 [M] (3), 477 [M−H] (7) 476 [M-2H] (12), 441 (18), 410 (10), 388 (14), 386 (20), 352 (19), 317 (6), 261 (7), 247 (7), 197 (6), 140 (12), 125 (100), and 89 (23).
4.5 2-((4-Hydroxyphenyl)(2-methoxybenzyl)amino)-5-((2-methoxy benzyl)amino)cyclohexa-2,5-diene-1,4-dione (C28H26N2O5) (4c)
Brown crystals, (75% yield); mp 185–186 C (dec.); 1H NMR (400 MHz, acetone-d6) δ 1.29 (s, 1H), 3.80 (s, 1H), 3.92 (s, 3H), 3.95 (s, 3H), 4.50 (d, J = 6.4 Hz, 2H), 5.27 (s, 1H), 5.97 (s, 2H), 6.55 (d, J = 7.8 Hz, 1H), 6.74 (t, J = 8.4 Hz, 2H), 6.88 (dd, J = 8.8, 2.5 Hz, 2H), 7.08 (t, J = 7.8 Hz, 3H), 7.31 (m, 4H), and 7.59 (d, J = 2.6 Hz, 1H) ppm; 13C NMR (125 MHz, DMSO-d6) δ 40.7, 42.6, 55.39, 55.41, 96.6, 104.7, 110.8, 112.0, 113.1, 115.8, 120.3, 124.4, 124.7, 125.8, 127.8, 128.3, 128.4, 132.3, 136.6, 149.6, 155.4, 156.2, 156.6, 176.8, and 178.1 ppm; IR (KBr): 3377, 3278, 3002, 2836, 1647, 1602, 1570, 1479, 1463, 1440, 1348, 1286, 1249, 1192, 1114, 1090, 1028, 950, 833, 796, and 754 cm−1; MS (EI): m/z (Relative intensity %): 468 [M-2H] (7), 392 (11), 378 (100), 364 (42), 347 (14), 271 (22), 257 (28), 136 (7), 121 (67), and 91 (98).
4.6 2,5-Bis(benzyl(methyl)amino)cyclohexa-2,5-diene-1,4-dione (C22H22N2O2) (7)
Light brown crystals, (94% yield); mp 151–152 C (dec.); 1H NMR (400 MHz, acetone-d6) δ 3.01 (s, 6H), 5.03 (s, 4H), 5.42 (s, 2H), 7.27 (d, J = 1.95 Hz, 2H), 7.28 (m, 6H), and 7.34 (d, J = 8.5 Hz, 4H). 13C NMR (125 MHz, DMSO-d6) δ 40.3, 56.1, 102.2, 126.8, 128.4, 137.4, 151.1, and 180.8 ppm; IR (KBr): 3060, 3030, 2912, 1711, 1623, 1563, 1494, 1452, 1411, 1358, 1338, 13132, 1215, 1098, 1027, 944, 866, 723, 692, and 464 cm−1; MS (EI): m/z (Relative intensity %): 346 [M] (63), 331 (32), 307 (21), 255 (83), 226 (9), 164 (11), 118 (13), 91 (100), and 65 (14).
Acknowledgments
We acknowledge the Bu-Ali Sina University Research Council and Center of Excellence in Development of Environmentally Friendly Methods for Chemical Synthesis (CEDEFMCS) for their support of this work.