

1 Introduction
In the last decades, intensive research studies were carried out to develop processes for the production of green, carbon-free hydrogen by water splitting [1,2]. This reaction can also be used for the characterization of reduced centers and oxygen vacancies of supported metal catalysts. High-temperature treatments of oxide-supported metal catalysts can create oxygen vacancies in the materials, particularly when these treatments are carried out in reducing media. Oxygen vacancies are thought to play a major role in catalysis and to be the real active sites of oxides in many instances [3].The most popular example is the reduction of titania in metal-TiO2 catalysts leading to the so-called SMSI effect (strong metal–support interactions) [4]. When a Pt-TiO2 is reduced at a low temperature (typically 300 °C), no effect is observed, the catalyst having a normal capacity to chemisorb hydrogen or carbon monoxide. On the contrary, when it is reduced at a high temperature (typically 500 °C), the chemisorptive properties of the catalyst are virtually suppressed [5]. Electronic effects may also explain the decrease of the catalytic activity of TiO2-supported metal catalysts (Pt, Ni) [6,7]. More recently, it has been shown that the control of the titania nanodomain size could be a way to modulate SMSI effects [8] while a treatment in an ultra-high vacuum and high temperature of Pt-TiO2 model catalysts could provoke the same SMSI phenomenon [9]. SMSI effects observed on metal-titania catalysts can be annihilated by treatment in air. Oxygen can re-oxide the TiO2−x moieties covering the metal and the titanium oxide thus formed comes back on the support [10]. This picture of the SMSI phenomenon was mainly established for the Pt-titania system. It seems less clear for Rh, even though this metal is mainly bonded to Ti3+ cations in the SMSI state [11]. If oxygen can annihilate the SMSI phenomenon by re-oxidation of the titanium suboxides, it seems that water may have a similar effect. The great advantage of water is that the metal itself is most probably not reoxidized. The reaction, analogous to water splitting, can be written as follows:
| 2Ti3+ + VO + H2O → 2 Ti4+ + O2− + H2(g) | (1) |
For several decades, ceria-based catalysts have been the subject of a huge number of studies [24–27]. Ceria is an oxide which can be reduced by hydrogen into a great variety of cerium suboxides CeO2-x with 0 < x < 0.5 [28]. The temperature-programmed reduction (TPR) profile of ceria generally consists of two main peaks: the first peak, ascribed to the reduction of the ceria surface, is centered around 450–500 °C while the second peak, starting at 550–600 °C corresponds to the reduction of the bulk of ceria [29–32]. When a noble metal is added to ceria, the first peak is shifted to lower temperatures (150–250 °C) while the high-temperature peaks is virtually unchanged [31,33,34]. In fact, the surface of ceria starts to reduce as soon as the noble metal itself is reduced. Substituting part of cerium with zirconium increases oxygen mobility and allows bulk reduction of the material at much lower temperatures than in pure ceria [35–37]. Although ceria can be easily reduced in the presence of metals, SMSI effects like those encountered with titania-supported catalysts are generally not observed: H2 and CO chemisorptions are virtually not affected by reduction at 500 °C [38] and HRTEM pictures show nanoparticles of metal (Rh, Pt …) not covered with CeO2-x moieties [17,38]. The modification of the catalytic properties of metal-ceria catalysts reduced at 500 °C are most often ascribed to electronic interactions, particularly for M-O-Ce sites located at the metal-ceria interface [27]. Metal nanoparticle decoration by CeO2−x moieties are observed only in catalyst samples reduced at very high temperatures (900 °C) [17,38]. In parallel, the stabilization of metal nanoparticles on reduced ceria could be of great interest for practical application [39]. The titration of O vacancies already used for Rh/TiO2 catalysts (Eq. (1)) can be applied to metal-supported ceria catalysts (Eq. (2)):
| 2Ce3+ + VO + H2O → 2 Ce4+ + O2− + H2(g) | (2) |
The objective of this paper is to review the main results obtained with selected metals (Rh or Pt) supported on alumina, silica, and ceria. In a preliminary section, the interaction of water with metallic surfaces will be reviewed to examine if water selectively reacts with O vacancies of the support or not.
2 Interaction of water with metallic surfaces
The behavior of water on metal surfaces was reviewed in 1988 by Heras and Viscido [40]. They have listed all the steps which may occur on metals, with the corresponding temperature range. The occurrence of these steps depends mainly on the energetics of OH, O and H adsorption and thus on the nature of metals and of their exposed faces:
| H2O (g) → H2O (ad) T < 100 K | (3) |
| H2O (ad) → H2O (g) T ≈ 200 K | (4) |
| H2O (ad) → OH (ad) + H(ad) 120 K < T < 200 K | (5) |
| 2 OH (ad) → H2O (g) + O (ad) 200 K < T < 350 K | (6) |
| OH (ad) → O (ad) + H (ad) T > 300 K | (7) |
| 2H (ad) → H2 (g) T > 300 K | (8) |
| O (ad) → O (bulk) T > 350 K | (9) |
| 2 O (ad) → O2 (g) T > 600 K | (10) |
On most metal surfaces, water is adsorbed at low temperatures (< 100 K) and can be dissociated as OH (ad) and H (ad) at higher temperatures. During temperature-programmed thermodesorption (TPD), water is evolved from the surface by recombination of OH groups (step 6). On certain metals (Fe, Co, Ni), the surface hydroxyl groups are irreversibly dissociated into O (ad) and H (ad) (step 7) and gaseous hydrogen can be formed by recombination of H (ad) species (Fig. 1). On clean Fe and Co films annealed at 77 K (−196 °C), TPD of adsorbed water exclusively leads to hydrogen while H2O and H2 are both desorbed from Ni surfaces [41]. When the films are annealed at higher temperatures, the ratio between H2 and H2O at the maximum desorption rate tends to decrease. The films are then less reactive for water dissociation. Oxygen pre-coverage dramatically changes the reactivity of water, favoring its dissociation on a number of metals. Hydroxyl groups formed upon H2O adsorption are stabilized by O atoms. Disproportionation of these OH groups reforms water desorbing at higher temperatures than on clean surfaces. In his review of 2002, Henderson confirms that irreversible dissociation of water occurs on Fe, Co, Ni, as well as on extremely reactive metals like Re, W, Al and Nb [42]. Hydrogen formation by reaction of H2O on Fe was utilized to quantitatively analyze the water formed during H2–O2 titrations on noble metal catalysts [43]. Water seems to be dissociated over Cu [44] and Ru [45] forming stable hydroxyl groups beyond 150 K [46] but hydrogen is not observed during H2O thermodesorption. At higher temperatures (350–500 K), water can be dissociatively adsorbed on polycrystalline copper with formation of hydrogen [47]. However, only 7–8% of the copper surface will readily dissociate water. On noble metals (Pt, Rh, Pd), water adsorption leads to clusters of H2O monomers, dimers; trimers,…, hexamers organized in bilayers on the clean metal surface [48–50]. On these noble metals, water is desorbed between 150 and 200 K. In neither case, hydrogen is detected during H2O thermodesorption. DFT calculations confirmed that intact water layers are more stable than dissociated water layers on these metals, except may be on Ru [51]. Oxygen pre-coverage may lead to H2O dissociation by reaction (11):
| O–Pt–Pt–H2O → OH–Pt–Pt–OH | (11) |
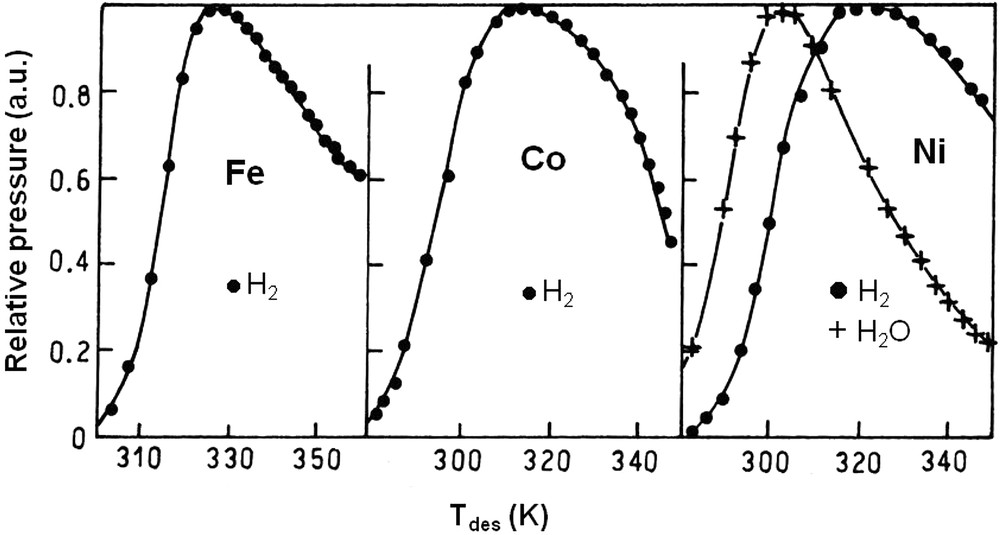
Thermal desorption spectra of water from clean surfaces of Fe, Co and Ni films annealed at 77 K. The ratio at the maximum desorption rate is infinite for Fe and Co (water desorbs as H2) and 1.8 for Ni. From ref. [41].
This has been shown in 1980 by Fisher and Sexton for Pt (111) [52] and confirmed later in numerous studies. Alloying Pt with Ru reinforces water dissociation in the gas phase but also in the liquid phase. Hydroxyl or oxygen species are then essentially coordinated to Ru atoms, which creates the conditions for a better CO oxidation activity in proton exchange membrane fuel cells designed for methanol fuel processing [53–55].
In conclusion, H2O is dissociated over Fe, Co and Ni. Thermodesorption of water leads exclusively to H2 (irreversible dissociation) over Fe and Co and partially to H2 over Ni. Water would be dissociated over Cu and Ru but the formation of H2 in the course of H2O thermodesorption is not clearly evidenced over these metals. Lastly, water dissociation would not occur over the most noble metals (Pt, Pd, Rh, Au) except over oxygen pre-covered metal surfaces. However, H2 is never observed during water TPD on these metals.
3 Reaction of water with alumina-supported metals
3.1 Role of water dissociation in steam reforming reactions
Interactions of water with supported metals were considerably less studied probably because adsorption of H2O on the support may largely bias the information which can be obtained by different techniques of characterization. The location of the sites (on the metal or on the support) where water can be adsorbed and dissociated plays an essential role in steam reforming reactions, even though electronic interactions and metal sites modification by the support cannot be excluded [56]. The catalytic behavior of metals supported on alumina or silica was compared for the toluene steam dealkylation reaction (12):
| C6H5-CH3 + 2H2O → C6H6 + CO2 + 3H2 | (12) |
This reaction can be viewed as a selective steam reforming reaction in which only the methyl group is gasified to produce benzene selectively [57]. On alumina, the relative turnover frequencies (TOF) of the metals at 440 °C are: Rh, 100 > Pd, 29 > Pt, 19 > Ni, 17 > Co, 15 > Ru, 14 > Ir, 13 [58]. Except for Ru, a similar ranking was found by Grenoble [59]. The reaction is not extremely sensitive to the nature of the metal since there is only one order of magnitude difference between the most active metal (Rh) and the less active one (Ir). By contrast, it was proven that the most active metals (Rh, Pt, Pd) were extremely support-sensitive. For instance, the TOF ratio between M/Al2O3 and M/SiO2 is 5.0 for Rh, 12.5 for Pt and 2.5 for Pd whereas Ni, Co, Ru and to a lesser extent Ir are virtually support-insensitive [57]. These results were explained by a change in the reaction mechanism depending on the nature of the active metal. Rh, Pt and Pd would not be able to dissociate the water molecule, this step occurring on the support. The better performances observed on alumina-supported metals are due to both a higher hydrophilicity and a higher mobility of the hydroxyl groups on Al2O3, necessary for the transport of OH reactive species from support to metal sites [60,61]. Support-insensitive metals would be able to dissociate water molecules so that all the steps occur on the metal (hydrocarbon and water activation and further reactions between intermediate species). This is in line with the results obtained on films and unsupported metals detailed in Section 2.
3.2 Water dissociation on alumina-supported metals (200–600 °C)
However, the presence of the support oxide and the nanosize of the metal clusters might change the reactivity with water. Therefore, we studied the decomposition of water on the alumina-supported metal catalysts listed in Table 1. The γ-Al2O3 was pretreated in H2 (16 h) at 900 °C prior to impregnation. The reaction was carried out in a pulsed chromatographic reactor as described in [62]. The catalyst sample was reduced in H2 at TR, outgassed in Ar (less than 1 ppm impurities) at the same temperature and then cooled down to TOX. Pulses of water (1 μl; 55.6 μmol) were vaporized and injected into the reduced catalyst. After trapping of the unreacted water, the hydrogen produced was analyzed by TCD. The results are qualitatively shown on Fig. 2. Metals can be ranked according to their reactivity with steam: Fe > Co >> Ru > Ni >> Ir >> Rh ≈ Pt ≈ Pd. The reactivity of Fe, Co and Ni on alumina is in agreement with the behavior of the unsupported metals (Section 2). Ru/Al2O3 is found more reactive than Ru films. At the opposite, Rh, Pt and Pd seems virtually inert with respect to water, minute amounts of H2 being recorded only at 600 °C.
Alumina-supported catalysts used for the reaction with pulses of H2O (see Fig. 3). Metal dispersions are measured by H chemisorption (Pd, Ni) or O titration of chemisorbed hydrogen (Rh, Pt, Ir) according to a procedure detailed in refs. [58] and [62].
| Metal | Rh | Pt | Pd | Ni | Ir | Co | Fe |
| Wt-% | 0.51 | 1.12 | 0.6 | 5.0 | 1.15 | 5 | 5 |
| Dispersion | 80 | 83 | 33 | 4.3 | 86 | n.d. | n.d. |
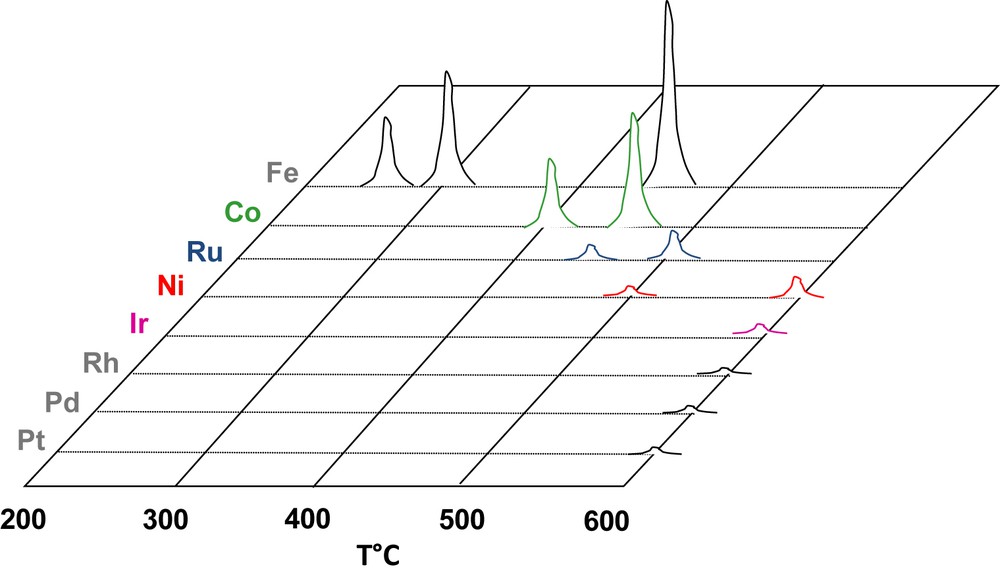
Thermal decomposition of pulses of water on alumina-supported catalysts.
3.3 Water dissociation on alumina-supported Rh and Pt (500–1000 °C)
High-temperature reduction of alumina-supported catalysts can lead to unusual catalytic properties, generally ascribed to metal sintering [63,64] and particle restructuration [65] or to encapsulation into alumina (evidenced for Rh [66] and Au [67]) or, merely, to a “special strong metal–support interaction” [68]. All these effects may occur during heat treatments but the formation of H2 by reaction of water is best explained by the presence of oxygen vacancies.
A detailed study was carried out on the alumina-supported Rh and Pt catalysts at higher temperatures of reduction (TR) and of reaction with steam (TOX) [69]. Hydrogen formation by decomposition of water on reduced catalysts is confirmed but hydrogen yields QH are significantly higher on Pt (15–30 μmol at H g−1) than on Rh catalysts (7–13 μmol at H g−1) (Table 2). After reaction with steam, the samples were characterized by H2 reduction at 500 °C. There is virtually no H2 uptake, which excludes the oxidation of the noble metal by steam. Hydrogen chemisorption and oxygen titration were also carried out at ambient temperature. They show that the metals are not deeply sintered, with dispersions of Rh and Pt close to 40% after reaction with steam at 850 °C.
Hydrogen formation (QH) by reaction of pulses of water (55.6 μmol) at TOX on alumina-supported Rh and Pt catalysts reduced at TR. From ref. [63].
| TR (°C) | TOX (°C) | QH (μmol at H g−1) | |
| Rh/Al2O3 | Pt/Al2O3 | ||
| 600 | 500 | 9.4 | 23.5 |
| 700 | 500 | 12.4 | 17.5 |
| 850 | 500 | 8.5 | 17.4 |
| 900 | 500 | 7.8 | 16.0 |
| 850 | 900 | 9.0 | 27.5 |
RhAl and PtAl alloys were prepared by arc melting Rh or Pt and Al powders under an Ar atmosphere in order to study their possible implication in the metal–support interaction following high-temperature reduction and their reactivity with steam. The RhAl alloy reacts easily with steam at 500°C–600 °C according to the following reaction:
| RhAl + 1.5H2O → Rh° + 0.5 Al2O3 + 1.5H2 | (13) |
The platinum-aluminum alloy is about two orders of magnitude less active than RhAl. Metallic platinum is not restored. Instead, a platinum-rich alloy, Pt5Al3, is formed. The reaction with steam thus produces hydrogen according to reaction 14:
| 5PtAl + 3H2O → Pt5Al3 + Al2O3 + 3H2 | (14) |
Reaction (15) leading to metallic platinum does not occur:
| PtAl + 1.5H2O → Pt° + 0.5 Al2O3 + 1.5H2 | (15) |
Steam reacts with reduced centers of alumina according to the following reaction:
| Reduced center + H2O → Oxidized center + H2 | (16) |
Although there is no absolute proof, the reacting centers are very likely located at the metal-alumina junction. The most convincing argument is that the bare support is totally inactive (QH ≈ 0) and that the reduced centers are created by a special interaction between the metal particles and the support. There are now more and more evidences that these sites can confer special physical properties to the materials [70,71] and play a major role in catalysis [72–74]. The number of Al–O moieties surrounding the metal particles can be estimated from the length of the metal–support interface. Details of the calculation are given in Appendix. At elevated temperatures of reduction, oxygen vacancies would be created at the metal–support interface according to the following reaction:
| O2− + H2 → ○2− + H2O | (17) |
Length of the metal–support interface I0 and theoretical amount of hydrogen QH* that can be formed by reaction of water on O vacancies located at this interface.
| Catalyst | Cubic particles | Hemispherical particles | ||
| I0/m g−1 | QH*/μmol H g−1 | I0/m g−1 | QH*/μmol H g−1 | |
| 0.51% Rh/Al2O3 | 3.5 × 108 | 3.4 | 7.3 × 108 | 7.0 |
| 1.12% Pt/Al2O3 | 4.1 × 108 | 3.9 | 8.5 × 108 | 8.2 |
4 Reaction of water with silica-supported metals
4.1 Formation of metal silicides by reduction of silica-supported metal catalysts
If the effects of high-temperature treatments on alumina-supported noble metals, in lean or rich atmosphere, were investigated in detail, possible effects on silica-supported catalysts were considerably less investigated. Changes in microstructures of Pt or Rh particles supported on silica were extensively investigated by Schmidt and coworkers in the 70–80's [77,78]. A sketch of the microstructure transformation in different atmospheres is shown on Fig. 3. High-temperature reduction can affect the particle morphology: platinum, for instance, tends to form cubes under H2. However, it seems that Schmidt and coworkers did not detect any special chemical interaction between the metal and the support, probably because the silica used in their works is a planar silica resistant to reactions with noble metals. On the contrary, metal silicides were detected in many instances on Ni/SiO2 [79], Pd/SiO2 [80–82], Pt/SiO2 [83–85] and Rh/SiO2 [23,86]. The main results of these studies are summarized in Table 4. Marot et al. showed that rhodium silicides could be formed by treatment of Rh-Si/SiOx films at 300 °C which means that Rh-Si alloying is relatively easy when a part of Si is already reduced [87,88]. Their results are also listed in Table 4. This strategy is widely used for producing silicide films at low temperatures [89]. Hippe et al. reported that Pt silicides (Pt3Si and Pt12Si5) were detected by electron diffraction in a 1.6 wt-%Pt/SiO2 (300 m2 g−1) reduced at 650 °C [85]. They observed that CO chemisorption, strongly annihilated on the catalyst reduced at 650 °C, could be partially restored by treatment under O2 at 350 °C followed by a reduction at 500 °C. It is likely that Pt silicides are less able than Pt to chemisorb CO. The catalyst deactivation is reinforced by the fact that Pt silicides tend to be enriched in silicon which cannot chemisorb CO. At the same time, Sadi et al. showed that rhodium silicides (Rh2Si) were formed by reduction of Rh/SiO2 catalysts (250 m2 g−1) at 600 °C and above [23]. To our knowledge, this is the only reference having studied the reactivity of these silicides with steam.
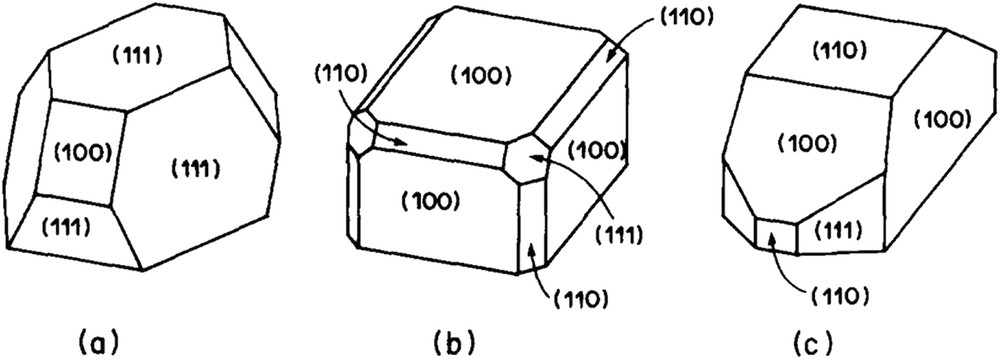
Microstructures of Pt or Rh particles on SiO2. The cuboctahedron (a) is formed by heating Pt in N2 or Rh in any gas. Heating Pt in H2 yields predominantly cubes (b) with face (100) parallel to the substrate surface, or truncated particles (c) exposing mainly (100) faces. From ref. [77].
Metal silicides detected in silica-supported catalysts or metal films/silica reduced at high temperatures.
| Catalyst or material | Support area m2 g−1 | Temperature range of reduction | Detected silicide | Ref. |
| 20%Ni | SiO2 Degussa 200 m2 g−1 | 910 °C | Ni–Si alloy (detected by magnetism) | [79] |
| 0.76%Pd | – | 600 °C | Pd3Si | [80] |
| Pd film 0.3 nm | SiO2 film 25–30 nm | 550–570 °C | Pd2Si | [81] |
| 10%Pd | SiO2 Davison 340 m2 g−1 | 450–500 °C | Pd3Si and Pd4Si | [82] |
| Pt film 3.2 nm | Quartz glass | 550 °C | Pt2Si and/or Pt3Si | [84] |
| Pt film 3 nm | Quartz | 550 °C (vacuum) | Pt3Si is suspected | [83] |
| 1.6%Pt | Sol–gel silica | 600–650 °C | Pt3Si and Pt12Si5 | [85] |
| 0.4 and 4%Rh | SiO2 RP 250 m2 g−1 | 500–980 °C | Rh2Si | [23] |
| Rh film (<1.5 nm) | SiO2 film (9 nm) evaporated on Mo foil | 600 °C | Rh3Si | [86] |
| Rh film | Si film (50–160 nm) on SiOx | Vacuum (XPS chamber) | Rh2Si (weak) | [87] |
| Rh film (50–140 nm) | Si film (50–160 nm) on SiOx | 300°C–900 °C (vacuum) | Rh2Si at 300 °C, Rh5Si3 at 500 and 600 °C, RhSi as well as Rh5Si3 at 800 and 900 °C | [88] |
4.2 Reaction of rhodium silicides with water
Two Rh catalysts were prepared (0.38 and 4.12 wt-% Rh) on a silica of 250 m2 g−1. They are denoted 0.4Rh/SiO2 and 4Rh/SiO2 respectively. Hydrogen chemisorption and oxygen titration performed after reduction at 400 °C show that these catalysts have close metal dispersion (12–13%) with a mean particle size of 6.8 ± 0.4 nm, confirmed by TEM. They were reduced for 1 h in H2 at different temperatures from 500 to 980 °C (samples denoted as 0.4 or 4Rh/SiO2RX where X(°C) is the reduction temperature TR; for instance 4Rh/SiO2R850 is the 4Rh/SiO2 catalyst reduced for 1 h at 850 °C). These reduced catalysts were finally characterized by reaction with pulses of H2O, also at different temperatures. The final sample is denoted as 0.4 or 4Rh/SiO2RXH2OY where Y is the temperature of reaction with steam TOX.
The results for TOX = 500 °C and TR from 500 to 900 °C are shown on Fig. 4. The curve corresponding to water titration of the prereduced 0.6%Rh/Al2O3 catalyst (Section 3) is reported for comparison. Contrary to what was observed on Rh/Al2O3, the amount of hydrogen formed by water titration on Rh/SiO2 significantly increases with the temperature of reduction. XRD and electron diffraction reveal that the rhodium silicide Rh2Si is formed according to the following reaction (written for 4Rh/SiO2; 4.12 wt-% or 2.46 mol%):
| 2.46Rh + 97.54SiO2 + 2.46H2 → 1.23Rh2Si + 96.31SiO2 + 2.46 + H2O | (18) |
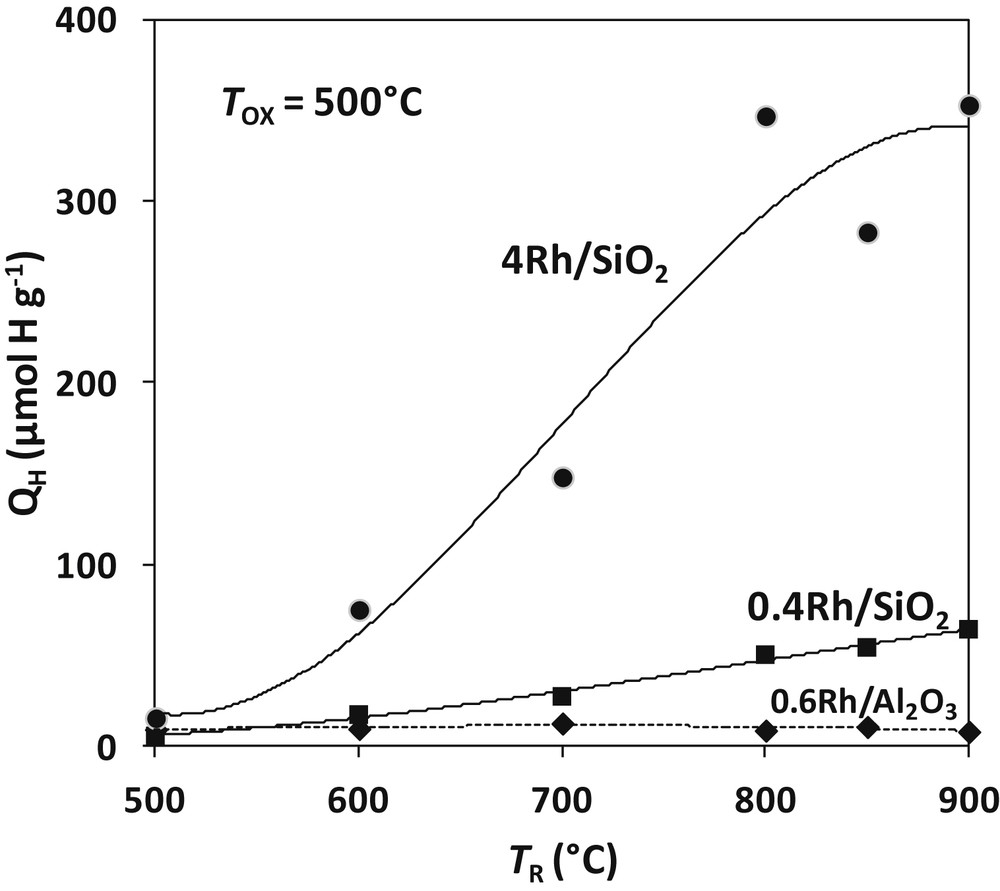
Amount of hydrogen formed by reaction of Rh/SiO2 catalysts prereduced at TR with pulses of H2O at 500 °C. The curve of the 0.6%Rh/Al2O3 (dotted line) is shown for comparison.
If all the rhodium was transformed into silicide, the production of H2 by titration QH (reverse reaction of 18) would be of 801 μmol H g−1 Fig. 4 shows that the highest H2 yield is 350 μmol H g−1: only 44% of the metal would react with silica to form the Rh2Si silicide. The same calculation gives 74 μmol H g−1 for the 0.4Rh/SiO2 catalyst, very close to the maximum value recorded after reduction at 900 °C (64 μmol H g−1; corresponding to 90% of the theoretical value of Rh2Si). The H2 yield varies slightly with the temperature of reaction TOX. QH amounts to 400 μmol H g−1 for 4Rh/SiO2 and 70 μmol H g−1 for 0.4Rh/SiO2 after reduction and reaction with water at 900 °C. It is confirmed that the silicide formation is limited on the 4Rh catalyst while it is almost total on the 0.4Rh sample.
4.3 Regeneration of the 4Rh/SiO2 catalyst after reduction at high temperatures
The 4Rh/SiO2 catalyst has been characterized by hydrogen chemisorption (HC) and cyclopentane hydrogenolysis at different stages of the pretreatment: (i) after reduction at TR, (ii) and then after reaction with pulses of H2O as described previously. The reaction of cyclopentane hydrogenolysis over Rh is a simple reaction leading mainly to n-pentane, even though a few amount of methane could be observed [90]: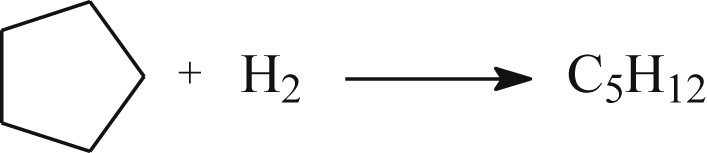
Pulses of cyclopentane (0.1 μl) were injected on the catalysts only prereduced at TR of first prereduced at TR and then reacted with pulses of water. Temperatures of reaction were varied from 170 to 270 °C. All these experiments were carried out in situ in the same chromatographic reactor described in [62]. The results are reported in Fig. 5. Hydrogenolysis activity at 200 °C are normalized with respect to the catalyst sample reduced at 500 °C. Hydrogen chemisorption values are also reported in Fig. 5. Both HC values and hydrogenolysis activity dramatically decrease when the catalyst is reduced at T ≥ 700 °C. Remarkably, chemisorption and catalytic activity are restored after the steam treatment, except for TR = 900 °C. Up to TR = 800 °C, the suppression of the catalytic activity is not due to metal sintering but silicide formation. At TR = 900 °C, the two phenomena (Rh sintering and Rh2Si formation) contribute to the catalyst deactivation. Evidently, H2O can regenerate the catalyst by alloy destruction but cannot redisperse the metal.
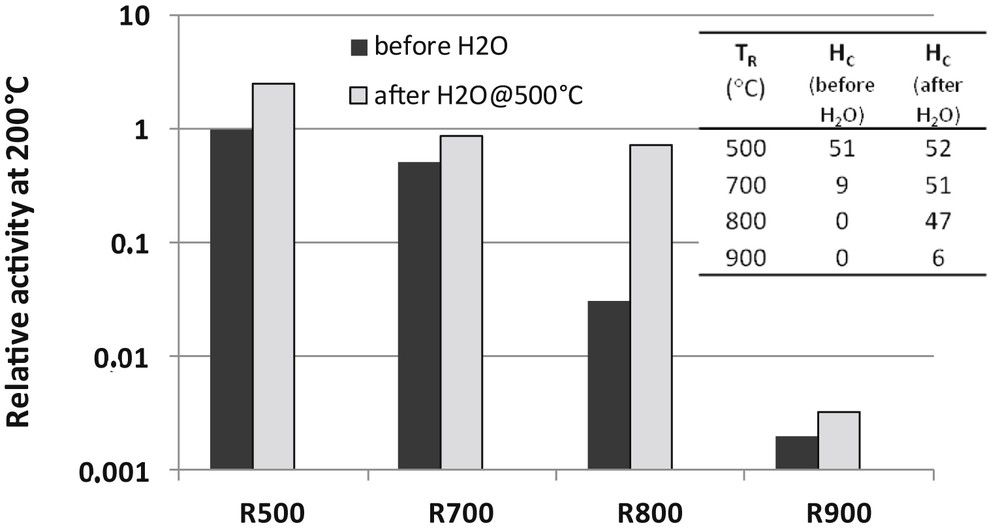
Effect of reduction temperatures and treatments in H2O at 500 °C on the relative activity of the 4Rh/SiO2 catalyst for the cyclopentane hydrogenolysis reaction at 200 °C. Activity after reduction at 500 °C is taken as unity. Values of hydrogen chemisorption after reduction and treatment in H2O (μmol H g−1) are given in inset.
5 Reaction of water with ceria-supported metals
5.1 Reduction by H2 of ceria-based catalysts
Ceria is a crucial component of three-way catalysts widely used for decades for its oxygen storage properties (OSC) [29]. Its redox properties combined with specific basic properties makes this oxide more and more applicable in oxidation reactions in the gas phase [91,92] or in water [93] and in organic synthesis reactions [25]. Ceria is a reducible support: Ce4+ ions are reduced by H2 into Ce3+ ions with correlative formation of oxygen vacancies VO:
| 2Ce4+ + 4O2− + H2 → 2Ce3+ + 3O2− + VO + H2O | (20) |
This equation is generally accepted even though DFT calculations show that the formation of Ce3+ does not imply necessarily the existence of oxygen vacancies [94]. Above 500 °C, the bulk of ceria can be reduced and deep reduction can be achieved between 500 and 1000 °C. Doping ceria with zirconia or praseodymia has a drastic effect on the reducibility [35]. Surface reduction is hardly increased but a significant bulk reduction can be observed at a medium temperature (300–400 °C) or at a relatively low temperature (200–250 °C) in the presence of metals. The very first centers of ceria are certainly difficult to be reduced: H adsorption and diffusion (but not H2O formation) would be easier on reduced ceria than on stoichiometric ceria [95].
Eq. (20) shows that one oxygen anion out of four can react with H2 to yield a water molecule, which corresponds to a consumption of 5.43 μmol H2 m−2 for surface reduction [93,96]. Zirconium ions being not reducible, the theoretical amount of H2 required for surface reduction of CexZr1−xO2 mixed oxides is μmol H2 g−1. The term takes into account the change in the O surface density with the Zr content [93,96]. The same equation holds for OSC inasmuch as oxygen storage capacity is most often measured by reduction of the oxide by pulses of CO which has the same oxygen-to-reductant stoichiometry as H2 [33].
5.2 Reaction of reduced ceria with H2O
Adsorption of water at low temperatures on ceria may lead to associative (H2O–H2O) or to dissociative adsorption (H–OH). This is a structure-sensitive process depending on the exposed face of ceria (100, 110 or 111), to the water coverage and to the initial state of ceria [94]. Reduced ceria CeO2−x is more able to dissociate water than the full oxide CeO2. Chen et al. investigated water adsorption on reduced CeO2 (111) surfaces and showed that dissociation (formation of hydroxyl groups) was irreversible at a temperature as low as 115K [97]. Thermodesorption leads to OH recombination but two competing reactions (Eqs. (21) and (22)) can occur:
| OH(a) + OH(a) → H2O(g) + Olattice + Ovacancy | (21) |
| OH(a) + OH(a) → H2(g) + 2Olattice | (22) |
Otsuka et al. were among the first to show that water could react with reduced ceria to produce hydrogen [98]. They also investigated the capability of ceria to convert CO2 into CO. Hydrogen is produced by the reverse reaction of Eq. (20) while CO is produced by reaction 23:
| 2Ce3+ + 3O2− + VO + CO2 → 2Ce4+ + 4O2- + CO | (23) |
The results obtained by Otsuka et al. on a ceria of 18.8 m2 g−1 are reported on Fig. 6. The reduction by H2 or CO is rather slow and should be carried out at 600 °C to get a percentage of reduction of 3.1%. But both reductants seem to reduce ceria at the same rate. By contrast, re-oxidation by H2O (H2 production) or by CO2 (CO production) are much easier. However, the re-oxidation by water (reaction at 300 °C) is faster than by CO2 (reaction at 400 °C). Otsuka et al. also showed that doping ceria with noble metals (Pt and Pd) did not significantly change the rate of reduction (factor 1.5–2) but considerably increased that of re-oxidation (factor 1000).
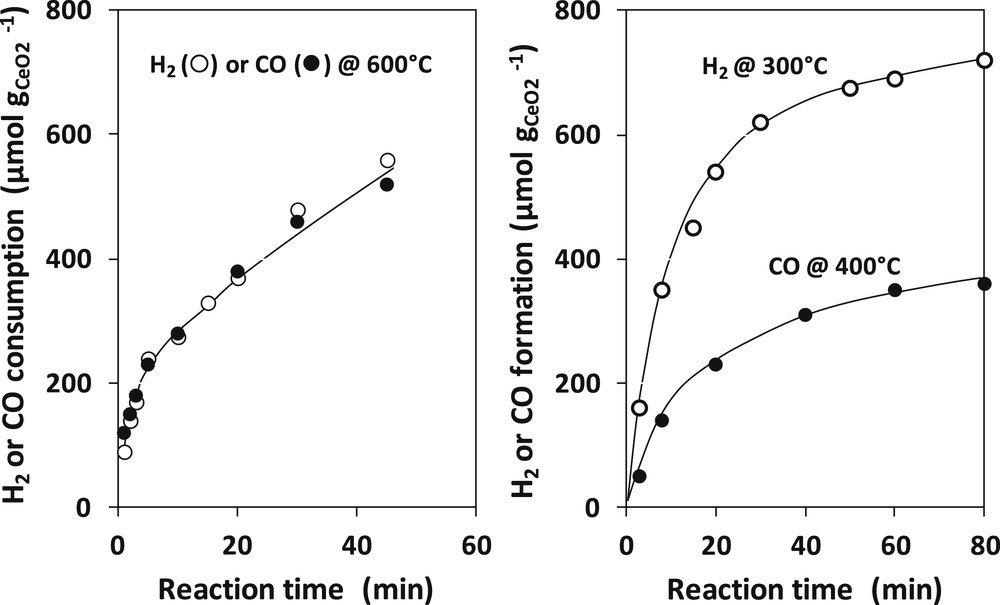
Kinetics of reduction of ceria by H2 and CO at 600 °C (left) and kinetics of re-oxidation by H2O at 300 °C and by CO2 at 400 °C (right). Adapted from Otsuka et al. [98].
Otsuka et al. used a commercial polycrystalline ceria. Recent investigations revealed that the reaction with H2O was structure-sensitive. Henderson et al. showed that water oxidation of Ce3+ ions can be observed on reduced ceria powder but not on a reduced CeO2 (111) surface [99]. They concluded that the reduced (111) surface is more resistant than non-(111) terminations for being oxidized by water. Nevertheless, the behavior differences between the (100), (110) and (111) faces strongly depend on the water pressure, which might explain the discrepancies observed between certain studies on this question [94,100].
The amount of H2 that can be produced by reaction of reduced ceria with H2O depends on the BET area of the material when it is reduced at medium temperatures (<500 °C). Obviously, when ceria is reduced at high temperatures (>900 °C), the totality of the bulk can participate in the hydrogen production. This can be achieved in solar thermochemical processes, in which temperatures of 1300–1500 °C can easily be reached. The use of a chemical reductant is then not necessary, since the oxide loses its oxygen at these temperatures. The main issues of these processes are to develop highly thermostable materials [101,102].
5.3 Reaction of reduced Pt or Rh/CeO2 with H2O
As recalled in Section 5.1, noble metals have a great impact on the surface reducibility of ceria but have little effect on its bulk reducibility. This is the reason why Otsuka et al. observed, at a high temperature (600 °C), a moderate increase in the rate of reduction of ceria (18 m2 g−1) in the presence of dopants (Pt, Pd) [98]. However, they reported that the metals were able to strongly accelerate the reaction of the reduced ceria with water. Lykhach et al. investigate the reactivity of water with model ceria(111) and Pt/ceria(111) catalysts in a low-temperature range (160–500K) [103]. They observed dissociation and re-oxidation by H2O of reduced ceria in the presence of Pt, but water was merely reformed by heating the sample above 250K. Kundakovic et al. reported water dissociation on a reduced Rh/CeO2 catalyst [104]. Contrary to Lykhach et al., hydrogen was observed during thermodesorption of water at 500K from reduced ceria and at 200–350K from reduced Rh/CeO2. Hydrogen formation is inhibited by the presence of coadsorbed CO.
More recently, Sadi et al. investigated the reactivity of steam on a 3.7% Rh/CeO2 (23 m2 g−1) reduced at TR ranging from 300 to 1000 °C [105]. Experimental conditions are similar to those used for alumina and silica catalysts: the catalyst sample is reduced for 1 h at TR, degassed at the same temperature in Ar. The reaction with water at TOX is carried out by pulses of 1 μl H2O. Hydrogen yields (QH in the logarithmic scale) for TOX = 500 °C are reported in Fig. 7 and compared to the values obtained with the 4Rh/SiO2 catalyst. QH values are about 8 times greater on Rh/CeO2 than on Rh/SiO2, the ratio between the two catalysts being independent of the temperature of reduction. Two theoretical values of QH should be considered: 262 μmol g−1 for a complete reduction of the surface of ceria and 5545 μmol g−1 for a complete reduction of the bulk of ceria. Fig. 7 shows that the surface reduction would be achieved by 600 °C in accordance with earlier TPR and spectroscopic studies [33,35,106]. The yield of hydrogen after 1 h reduction at 850 °C (2795 μmol g−1) would correspond to a bulk reduction of 50.4%. Keeping constant the temperature of reduction (850 °C), the temperature of re-oxidation TOX was varied between 100 and 850 °C in order to examine the reactivity of reduced Rh/CeO2 with water. The results reported in Table 5 show a significant reactivity of water at low temperatures. At 100 °C, 74% of the reduced sites are titrated by H2O. The metal plays a major role in this high reactivity: reaction of water with reduced ceria generally occurs at much higher temperatures (>600 °C) [101]. The question whether H2 could originate from some hydrogen stored in the material during reduction also arose. The problem was elucidated by experiments in which reduction was carried out in D2 and reaction with H2O. Negligible amounts of D2 were obtained, which confirmed that H2 was produced by water splitting [105]. At highest temperatures (>850 °C), rhodium cerides (CeRh and Ce5Rh4) were clearly identified by XRD and electron diffraction. Metallic rhodium only was found in the catalysts after re-oxidation by H2O. Reaction 24 (written with RhCe) similar to reaction 18 with silicides seems to occur with cerides:
| RhCe + H2O → Rh + CeO2+2H2 | (24) |
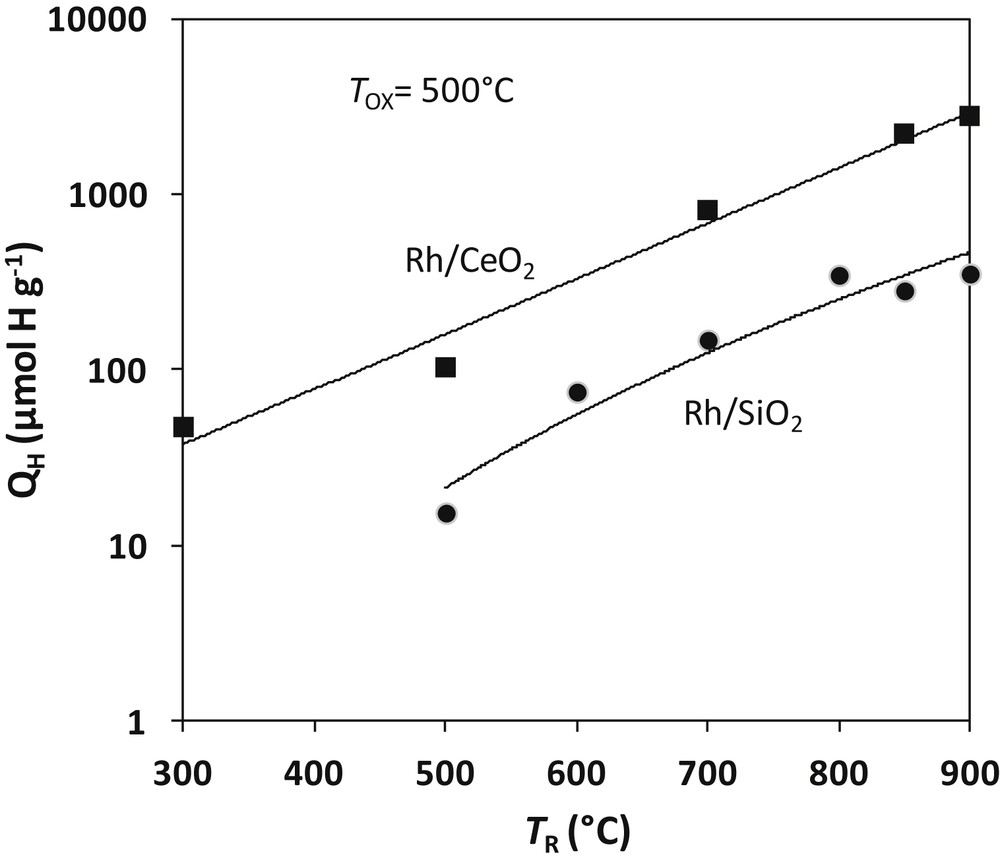
Water splitting on reduced Rh/CeO2 and Rh/SiO2 catalysts. Amount of hydrogen produced by reaction of water on reduced 3.7 wt-%Rh/CeO2 compared to the 4.12%Rh/SiO2 of Fig. 5. Adapted from Sadi et al. [105].
Effect of the temperature TOX on the re-oxidation by water of a 3.7 wt-% Rh/CeO2 reduced for 1 h at 850 °C. R% is the percentage of reduction based on QH = 5545 μmol g−1 for a total reduction of the bulk of ceria. The corresponding general formula is CeO2−x with x = R/200. From Sadi et al. [105].
| TOX (°C) | QH (μmol H g−1) | R% | General formula |
| 100 | 2064 | 37.2 | CeO1.814 |
| 500 | 2213 | 39.9 | CeO1.800 |
| 700 | 2685 | 48.4 | CeO1.758 |
| 850 | 2795 | 50.4 | CeO1.748 |
In this case again, the reaction with water allows us to selectively titrate the reduced centers of the support.
6 Conclusions
The reaction of H2O on reduced metal catalysts leads to formation of H2 and filling of oxygen vacancies by water splitting. The use of pulses of water is recommended for a good sensitivity of H2 detection. Noble metals (Rh and Pt in many studies) should be used to avoid the metal re-oxidation by water. Surface science investigations showed that water can be dissociated over Fe, Co, Ni, Ru, Cu with total or partial formation of H2. Three cases were examined in this review:
- 1– Case #1. Pt and Rh/Al2O3 were reduced at temperatures ranging from 600 to 900 °C. The amount QH of hydrogen formed by water splitting is coherent with the presence of oxygen vacancies located at the metal periphery. The formation of O vacancies without reduction of Al3+ ions is discussed but the formation of RhAl or PtAl moieties is not excluded.
- 2– Case #2. The reduction at TR > 600 °C of metal/SiO2 catalysts leads to the formation of metal silicides such as Rh2Si. Evidence of silicide formation is reported in many studies, particularly with Pt and Rh catalysts. The reaction of water at TOX > 600 °C destroys the silicide by selective re-oxidation of Si. Hydrogen chemisorption and rate of cyclopentane hydrogenolysis strongly decrease when the silicide is formed. Initial properties of the catalyst are restored after reaction with water.
- 3– Case #3. Surface reduction of ceria is detected above 200 °C. At TR > 600 °C, the bulk of ceria starts to reduce. The amount of hydrogen formed by reaction with water shows that 50% of ceria is reduced at 850 °C, which corresponds to a mean composition of Rh/CeO1.75. Reaction of water with reduced Rh/CeO2 is significant at low temperatures: at TOX = 100 °C, 75% of the maximal amount of H2 can be formed. When the temperature of reduction exceeds 900 °C, rhodium cerides are detected in the catalyst by XRD and electron diffraction.
In these studies, the water splitting reaction was carried out by means of pulses of water. This technique allows the detection of very small amounts of hydrogen, estimated at less than 1 μmol g−1. This should be compared to the number of H2O molecules required for titrating the oxygen vacancies in a 1 %Pt/Al2O3 (about 10 μmol eq.H g−1) and, at the opposite, to the high number of oxygen vacancies at the surface of ceria (262 μmol g−1 for a BET area of 23 m2 g−1 in case #3). In this latter case, the technique is able to measure variations of less than 1% of the number of vacancies at the surface of ceria.
Appendix Catalyst characteristics: particle size, area, number of particles, length of the metal–support interface.
Let us consider the following parameters for the catalyst:
- - metal content: xm (wt-%)
- - dispersion: D%
- - particle size: d (m)
- - number of particles per gram: N
and for the metal:
- - molar weight: Mm (g mol−1)
- - density: ρ (g m−3)
- - intrinsic surface area for one monolayer of the metal: σ (m2 mol−1)
For one cubic nanoparticle (sitting on one face), the surface area is a = 5 d2 and the weight of the metal is m = ρd3.
For 1 g of catalyst:
- - metal area (m2 ):
| (A-1) |
- - weight of metal (g metal ):
| (A-2) |
- - length of the metal–support interface (m ):
| (A-3) |
The particle size and the number of particles can be deduced by combining Eqs. A-1 and A-2:
| (A-4) |
| (A-5) |
| (A-6) |
For Rh catalysts (Mm = 102.9 g mol−1; ρ = 12.45 × 106 g m−3 and σ = 47,630 m2 mol−1), Eqs. A-4, A-5 and A-6 give:
| (A-7) |
while for Pt (Mm = 195.1 g mol−1; ρ = 21.09 × 106 g m−3 and σ = 50710 m2 mol−1), we have:
| (A-8) |
For hemispherical nanoparticles (sitting on a flat circular face), the same calculation leads to:
| (A-9) |
| (A-10) |
| (A-11) |
For Rh catalysts with hemispherical particles, one has:
| (A-12) |
and for Pt catalysts:
| (A-13) |