

1 Introduction
Les composés organiques volatils (COV) sont des composés toxiques et/ou précurseurs d’ozone, qui sont libérés dans l’atmosphère à température ambiante et contribuent de cette manière à la pollution de l’air [1,2]. Réduire leur émission dans l’atmosphère constitue une préoccupation environnementale et un réel enjeu de santé publique. Plusieurs procédés ont été mis en place pour limiter les émissions de COV dans l’atmosphère (biofiltres, oxydation thermique ou catalytique) [2]. Parmi ces méthodes, l’oxydation catalytique reste une technique de choix pour l’élimination, à faibles concentrations, des COV. Toutefois, l’efficacité de cette méthode dépend essentiellement de catalyseurs efficaces qui présentent une surface spécifique élevée, une bonne stabilité thermique ainsi que de bonnes propriétés oxydo-réductrices.
Plusieurs études ont été réalisées sur l’oxydation catalytique des composés organiques volatils [3–6]. L'élimination de polluants tels que le toluène, le propène ou encore le butanol a déjà été discutée dans la littérature. Cependant, les recherches concernant l’acide acétique en font rarement référence comme COV, bien qu’il soit nocif pour la santé humaine. En effet, l’acide acétique est plutôt étudié en oxydation par voie humide et considéré comme la molécule la plus réfractaire à l’oxydation d’un grand nombre de composés [7–10]. Il est présent dans l'atmosphère, car il est susceptible d'être émis par plusieurs végétaux dont il constitue la sève. Il est également émis de manière naturelle par l'homme à travers les urines, la sueur, le lait, etc. D'autres sources d'émission de l'acide acétique dans l'atmosphère proviennent des gaz d'échappement des véhicules et de la combustion de végétaux, de plastiques ou d'autres déchets [11]. L'acide acétique pur est une substance dangereuse, très corrosive et irritante. C'est un composé stable, peu dégradable mais capable de se décomposer à 442 °C en méthane, dioxyde de carbone, cétène et eau. L’oxydation voie humide catalysée (OVHC) est une technique d’élimination de polluants industriels en phase aqueuse. Elle consiste à oxyder les effluents gazeux (phénol, acide acétique…) à pression et température basses. L’OVHC de l’acide acétique a fait l’objet de plusieurs études dans la communauté scientifique. En effet, l’acide acétique est connu comme étant un intermédiaire réactionnel dans les réactions d’oxydation des composés organiques telles que l’oxydation du phénol [7].
Lors de l’oxydation par voie humide catalysée (OVHC), l’acide acétique formé en phase aqueuse peut facilement, puisque volatil, se retrouver dans la phase gazeuse. Ainsi, l’objectif de ce travail est d’utiliser des matériaux catalytiques novateurs et peu onéreux afin que leurs propriétés acido-basiques et/ou redox aident à l’oxydation totale de l’acide acétique en phase gazeuse.
Les catalyseurs choisis sont des mélanges d'oxydes à base de magnésium (Mg) et d'aluminium (Al), issus de la famille des hydrotalcites, sur lesquels un dépôt de cérium a été réalisé afin d'améliorer leur réductibilité. En effet, la littérature montre que, selon le mode de préparation et/ou par suite de l’ajout d’un métal réducteur fort tel que le cérium, il est possible d’améliorer les performances des catalyseurs [4,12]. De la même manière, les oxydes mixtes MgAl(O) obtenus après calcination d’hydrotalcites précurseurs MgAl possèdent une grande surface spécifique capable d'améliorer l'activité des catalyseurs, une forte basicité susceptible de favoriser l’adsorption de l’acide acétique et une résistance thermique élevée. Dans ce travail, une série de catalyseurs MgAlCey (y : pourcentage molaire de cérium), préparés par co-précipitation et par voie sol–gel puis caractérisés par différentes techniques ont été testés dans l'oxydation de l’acide acétique à basse température (T ≤ 500 °C) afin de déterminer leurs performances catalytiques.
2 Partie expérimentale
2.1 Préparation des catalyseurs
2.1.1 Co-précipitation
Le catalyseur obtenu par co-précipitation a été préparé à partir d’une solution (1 mol.L−1) de précurseurs de nitrates de magnésium (Mg(NO3)2·6H2O) (MERCK) et d’aluminium (Al(NO3)3·9H2O) (MERCK) dissous goutte à goutte dans une solution basique NaOH (1 mol.L−1)/Na2CO3 (0,5 mol.L−1) (SIGMA–ALDRICH). Le mélange est maintenu à pH ≥ 10 sous agitation pendant une nuit à 80 °C. Le précipité obtenu est filtré, lavé plusieurs fois à l’eau distillée, séché à l’étuve à 100 °C pendant 24 h, puis calciné dans un four à moufle à 500 °C pendant 4 h (2 °C/min). Après calcination, le solide homogène Mg(1−x)Al(x)(O) obtenu est nommé MgAl_cp (avec x = 0,25 et ratio molaire Mg/Al = 3). Le catalyseur MgAlCey_cp (y = 0,14) est préparé par imprégnation voie humide à partir d’une solution colloïdale éthanolique d’acétate de cérium sesquihydraté Ce(C2H3O2)3·5H2O et des précurseurs de la phase MgAl. Le mélange reste sous agitation vigoureuse pendant 1h30, puis est séché sur un banc de sable à 80 °C et calciné à 500 °C pendant 4 h. La teneur métallique en cérium est vérifiée par spectrométrie d’émission optique à plasma inductif (ICP-OES).
2.1.2 Sol–gel
Le catalyseur Mg(1−x)Al(x)(O), nommé MgAl_sg (avec x = 0,25 et ratio molaire Mg/Al = 3), est également synthétisé par la méthode sol–gel. Le précurseur éthoxyde de magnésium (C4H10MgO2) est dissout dans un volume d’éthanol nécessaire (rapport molaire solvant/précurseur = 17). Le mélange est chauffé à reflux sous agitation vigoureuse jusqu’à 70 °C. Après 15 min d’agitation, quelques gouttes d’acide nitrique sont ajoutées pour favoriser la condensation du gel. Lorsque le pH est proche de 5, la solution obtenue est maintenue sous agitation pendant 2h30. Après cette étape, le tri-sec-butoxide d’aluminium (Al(C4H9O)3) est additionné au mélange accompagné d’un volume d’eau ultra pure approprié (nsolvant/nprécurseur égal à 100). L’agitation est maintenue pendant 15 min à 350 tr.min−1. Le gel obtenu est ensuite séché sur un banc de sable à 80 °C, puis calciné à 500 °C pendant 4 h. Le catalyseur MgAlCey_sg (y = 0,14) est préparé par imprégnation voie humide, dans les mêmes conditions que le matériau obtenu en co-précipitation et sa teneur en cérium est également vérifiée par analyse ICP-OES.
La cérine (CeO2) est obtenue par calcination d’acétate de cérium sesquihydraté à 500 °C pendant 4 h.
2.2 Caractérisations des catalyseurs
2.2.1 Surface spécifique (BET)
Les surfaces spécifiques des catalyseurs sont mesurées selon la méthode BET (Brunauer–Emmet–Teller) à l'aide d'un appareil Micromeritics TriStar 3000. Environ 200 mg de poudre sont prétraités sous vide, dans une cellule spécifique, pendant une nuit à 250 °C. Après analyse, les surfaces spécifiques sont déduites des isothermes d'adsorption–désorption de N2 à 77 K.
2.2.2 Capacité de stockage en oxygène (CSO)
La capacité de stockage en oxygène correspond à la quantité d'oxygène immédiatement disponible sur l'échantillon. La technique utilisée consiste à suivre, en régime pulsé de monoxyde de carbone, la réduction à 400 °C par le CO des échantillons pré-calcinés à 500 °C sous O2 [4]. Le CO2 formé est purgé par un flux d'hélium (30 mL.min−1), puis analysé en sortie du réacteur via un système chromatographique (détecteur TCD). Après un calcul numérique, la valeur obtenue, nommée CSO, représente la capacité de stockage en oxygène.
2.2.3 Chimisorption du CO2
La chimisorption du CO2 est une technique qui permet de mesurer la basicité des échantillons à l’aide d’un montage de chromatographie pulsée. L’échantillon est préalablement traité sous flux d'hélium (30 mL.min−1) à 500 °C pendant 15 min, puis des pulses de CO2 sont injectés toutes les 3 min jusqu’à saturation de la surface. La quantité de sites basiques correspond ainsi à la proportion du CO2 chimisorbé sur le matériau.
2.2.4 Adsorption du CO2 suivie par spectrométrie infrarouge à transformée de Fourier (FTIR-CO2)
Le dioxyde de carbone est utilisé comme molécule sonde pour caractériser la basicité de surface des matériaux par l'identification des espèces carbonates formées. Le catalyseur est compressé sous 2 tonnes.cm−2 à l’aide d’une presse hydraulique et la pastille formée (20–30 mg), de diamètre 16 mm et de surface 2 cm2, est prétraitée sous vide à 450 °C pendant une nuit. L’adsorption du CO2 est réalisée en saturant la pastille durant 30 min sous une pression de 25 mbar. Les spectres infrarouge, normalisés à 10 mg, sont enregistrés à 24 °C, 150 °C, 250 °C, 350 °C et 450 °C à l’aide du spectrophotomètre Nicolet 5700 à transformée de Fourier. La thermo-désorption du CO2, enregistrée dans la région 1800–1200 cm−1, permet d’évaluer la force des sites basiques. Le nombre de sites basiques identifiés est obtenu à température ambiante (24 °C) par la relation suivante:
2.3 Test catalytique
Les performances catalytiques des matériaux sont évaluées au cours de la réaction d’oxydation de l’acide acétique. Cette réaction est réalisée sur la base du protocole utilisé par Sedjame et al. [4], à l’aide d’un réacteur tubulaire à lit fixe, selon les conditions suivantes : mélange gazeux air synthétique-polluant contenant 20% d’O2, 76,9% de N2, 3% d’H2O et 0,1% de COV avec une masse de catalyseur fixée à 140 mg, diluée dans 1 g de cordiérite. Le flux total est de 70 mL.min−1. Les tests sont effectués entre 100 °C et 500 °C. En effet, les gaz sont préalablement envoyés dans le réacteur à T = 100 °C pendant 1h30, puis, une montée de température est mise en place jusqu’à 500 °C avec une rampe de 3 °C.min−1. Les produits de réaction sont analysés par micro-chromatographie en phase gazeuse pilotée par un logiciel intégré (Galaxie Chromatographie). Les formules suivantes permettent le calcul de la conversion du réactif et du rendement en produits:
3 Résultats et discussions
3.1 Influence de la cérine à la surface des matériaux
Les résultats d’analyse de surface spécifique (BET) des matériaux sont représentés dans le Tableau 1 et montrent que les solides sont mésoporeux. Les oxydes mixtes MgAl_cp et MgAl_sg, synthétisés par les deux méthodes, possèdent une surface spécifique élevée (SBET ≥ 150 m2.g−1). Les traitements des catalyseurs par calcination induisent des phénomènes de déshydroxylation, de déshydratation et de décarboxylation [13,14] à l’origine de la formation de lacunes anioniques dans la structure, ce qui augmente la porosité des matériaux et leur confère une surface spécifique élevée. De plus, la surface spécifique et le volume poreux du catalyseur MgAl sont nettement plus importante lorsque celui-ci est préparé par co-précipitation plutôt que par voie sol–gel à cause de la formation d’espaces inter-feuillets via l’intercalation d’anions carbonate (
Propriétés physico-chimiques des échantillons synthétisés par co-précipitation (cp) et sol–gel (sg). Taille des cristallites calculée d'après la raie du plan (022) des diffractogrammes commune à tous les matériaux.
| Échantillons | S.SBET (m2.g−1) | Volume poreux (cm3.g−1) | Diamètre des pores (nm) | CSO (μmol.gcat−1) | Nombre de sites basiques (μmol.m−2) | Taille des cristallites (nm) | ||||||
| cp | sg | cp | sg | cp | sg | cp | sg | cp | sg | cp | sg | |
| MgAl | 276 | 218 | 0,76 | 0,41 | 10,9 | 7,52 | 0 | 8 | 0,3 | 0,9 | 3,8 | 4,3 |
| MgAlCe0,14 | 164 | 175 | 0,38 | 0,33 | 9,27 | 7,52 | 156 | 129 | 0,5 | 1,8 | 3,5 | 5,5 |
| CeO2 | 72 | 0,10 | 5,65 | 137 | 1 | 8,5 |

Diffractogrammes des matériaux synthétisés obtenus après calcination.
Les capacités de stockage d’oxygène de l’ensemble des matériaux synthétisés sont présentées dans le Tableau 1. Les résultats obtenus montrent que l’ajout de la phase active CeO2 augmente la capacité de stockage des solides. Pour l’échantillon MgAlCe0,14_cp, l’ajout de cérium fait croître la CSO jusqu’à 156 μmol.gcat−1, et un effet de synergie est observé. La CSO, exprimée par gramme de catalyseur, est plus forte pour le matériau dopé que pour la cérine pure. Pour l’échantillon MgAlCe0,14_sg, la présence de cérine influence également la CSO. La valeur obtenue est plus faible que pour le matériau obtenu par co-précipitation (129 μmol.gcat−1). La CSO représente l'oxygène réactif susceptible d'être stocké ou libéré par le catalyseur. La mobilité de surface de cet oxygène est donc directement liée aux propriétés redox du matériau. Martin et Duprez [19,20] ont démontré que la réductibilité de la cérine est due à la présence de différentes espèces d’oxygène (
Lorsque l’oxygène, disponible au sein du matériau réductible, provient de la première couche d’oxyde de cérium, NC ≤ 1. Dans le cas où NC ≥ 1, l’oxygène transféré provient de plusieurs couches. Les matériaux MgAlCe0,14_cp et MgAlCe0,14_sg possèdent respectivement un nombre de couches égal à 1,91 et 1,21. Ces catalyseurs présentent des valeurs de NC ≥ 1, ce qui signifie que plusieurs couches de cérium sont impliquées dans le transfert de l’oxygène. Une légère différence existe malgré tout en fonction du type de matériau, car le nombre de couches impliquées dans l’échange d’oxygène est plus élevé en co-précipitation qu’en sol–gel. Ce résultat traduit simplement le fait que l’oxygène de surface est en plus grand nombre et plus facilement accessible sur le catalyseur MgAlCe0,14_cp. De la même manière, la dispersion des particules de cérium est meilleure à la surface du catalyseur préparé par co-précipitation plutôt qu’en sol–gel du fait de la plus petite taille des particules. Par conséquent, la présence de cérine de surface, bien dispersée et accessible, favorise sa réductibilité lors de la mesure de CSO.
La quantité totale de sites basiques des catalyseurs est indiquée dans le Tableau 1. Il apparaît clairement que le catalyseur MgAlCe0,14_cp est plus basique que l’échantillon MgAlCe0,14_sg. De toute évidence, la présence de cérium augmente le nombre de sites basiques des catalyseurs, alors qu’un effet de synergie est constaté sur MgAlCe0,14_cp. Les catalyseurs MgAl_cp et MgAlCe0,14_cp possèdent le plus grand nombre de sites basiques. En effet, les études menées par Bolognini et al. [23] ont montré que les propriétés basiques des oxydes mixtes de type MgAl diffèrent entre les deux modes de préparation (co-précipitation et sol–gel). En général, pour un même rapport Mg/Al et comme le confirment nos résultats, le nombre de sites basiques des échantillons synthétisés par voie sol–gel est inférieur à celui des catalyseurs obtenus par co-précipitation. Cette différence est probablement due à la présence d'une quantité de sites basiques moyens (Mg2+–O2−) supérieure lorsque le matériau est synthétisé par co-précipitation. Lavalley et al. [24] ont montré que l’adsorption du CO2 à la surface des oxydes métalliques génère différentes espèces de carbonates. Ces carbonates caractérisent la force de basicité des matériaux. Les sites faibles sont formés à partir des groupements hydroxy (OH) à la surface du catalyseur tandis les sites moyens et forts sont dus aux ions O2− dont la coordination dépend de la liaison métal-oxygène Mn+–O−.
Les analyses infrarouges préalablement réalisées à température ambiante (24 °C) ont permis l’identification des espèces carbonate formées à la surface. À partir des aires de bandes spectrales, le nombre et la force de chaque site sont déterminés sur les catalyseurs CeO2, MgAl et MgAlCe0,14 préparés par voie sol–gel et par co-précipitation. Les résultats obtenus sur les catalyseurs frais (avant test catalytique) CeO2, MgAl_cp et MgAl_sg sont représentés sur la Fig. 2. Le catalyseur MgAl frais possède, effectivement, un nombre de sites basiques moyens plus élevés en co-précipitation qu’en sol–gel. De plus, ces sites sont majoritairement formés à la surface du catalyseur MgAl_cp. Cependant, les sites faibles OH sont plus élevés en sol–gel qu’en co-précipitation, alors que le contraire est observé pour les sites forts O2−. La contribution globale des différentes forces des sites de surface semble indiquer que la force basique du matériau MgAl_cp est supérieure à celle de MgAl_sg. Quant à la cérine, la basicité est due à un nombre élevé de sites moyens et forts. En effet, selon plusieurs auteurs [25], la formation de carbonates à la surface de la cérine dépend essentiellement des liaisons métal-oxygène Mn+–O−, lesquelles favorisent la formation des sites moyens et forts. Toutefois, l'hétérogénéité de la surface des oxydes mixtes MgAlCe0,14 met en évidence la présence de cations Ce4+ et Al3+ liés à l'existence d'autres sites basiques. En effet, selon le modèle de Knözinger et Ratnasamy [26], les cations Al3+ des sites tétraédriques de la structure MgAl sont responsables de l'apparition des groupements hydroxy OH liés à une faible basicité [26] au sein des matériaux. Quant aux cations Ce4+, ils occupent la moitié des sites interstitiels de la maille cubique simple de la structure fluorine de CeO2, avec comme voisins des anions oxygène caractéristiques d'une basicité élevée [20,27]. En général, la basicité des catalyseurs est en lien avec le nombre d’espèces métalliques Mg2+, Al3+ et Ce4+de surface [19] et les liaisons métal–oxygène Mn+–O− [28]. Les résultats de l’analyse infrarouge obtenus sur les catalyseurs frais et usés (après test catalytique) MgAlCe0,14 sont représentés sur la Fig. 3. Il apparaît, sur la Fig. 3a, que le nombre de sites forts du catalyseur frais MgAlCe0,14 est plus élevé en sol–gel qu’en co-précipitation. Le contraire est observé sur les sites moyens (Mg2+–O2−) à la surface des catalyseurs puisque les valeurs sont plus élevées en sol–gel qu’en co-précipitation. Il n’y a pas de grande différence entre les sites faibles des catalyseurs frais MgAlCe0,14_cp et MgAlCe0,14_sg. En effet, la présence de cérine à la surface des catalyseurs favorise la formation des sites moyens (Mg2+–O2−) et forts O2−. Il est probable que lorsque la cérine se dépose à la surface des catalyseurs, le recouvrement des sites basiques par les particules d’oxyde de cérium se fait préférentiellement sur les sites moyens (Mg2+–O2−) du matériau catalytique MgAl_cp et sur les sites faibles Al3+–O− du catalyseur MgAl_sg. Une partie des sites basiques faibles et moyens est effectivement remplacée par les sites basiques forts O2− de la cérine, de telle sorte qu'en surface, le nombre des sites basiques moyens (Mg2+–O2−) et faibles Al3+–O− diminue. Cependant, le recouvrement des sites du catalyseur MgAlCe0,14_sg crée une augmentation des sites basiques moyens. Sur le catalyseur MgAlCe0,14_cp, la formation des sites forts est améliorée. Binet et al. [25] ont montré que les sites moyens de la cérine correspondent à la formation d’espèces carbonatées pontées ou bidentées et que les espèces monodentées ou polydentées caractérisent les sites forts. Leur interaction à la surface des catalyseurs dépend de la coordination de la liaison métal–oxygène Mn+–O−. La géométrie précise de cette interaction ne pouvant pas être réalisée, il semble donc que les espèces carbonatées pontées ou bidentées sont favorisées à la surface du catalyseur MgAlCe0,14_sg et les espèces monodentées ou polydentées sont privilégiées en co-précipitation. De toute évidence, la présence de cérium à la surface des catalyseurs améliore la basicité des matériaux. Sur la Fig. 3b sont représentés les résultats de l’analyse infrarouge des catalyseurs MgAlCe0,14_cp et MgAlCe0,14_sg usés après un cycle de conversion de l’acide acétique. Il est intéressant de constater que la contribution en surface des sites basiques des catalyseurs change après la réaction d’oxydation. Le nombre de sites basiques forts du catalyseur usé MgAlCe0,14 diminue, pendant qu’un enrichissement des autres sites s’opère à la surface. En effet, en co-précipitation, le nombre de sites faibles et moyens a augmenté et, en sol–gel, les sites basiques moyens sont les seuls à augmenter. D’après Calaza et al. [29], les transformations de l’acide acétique à la surface de la cérine affectent la disponibilité de l’oxygène car ce dernier permet la stabilisation et la modification de l’adsorbat. De ce fait, cette modification entraîne un changement de la coordination de la liaison métal–oxygène Mn+–O− et favorise la formation des autres sites.

Quantification des sites basiques faibles, moyens et forts des catalyseurs frais CeO2 et MgAl préparés par voie sol–gel et par co-précipitation.
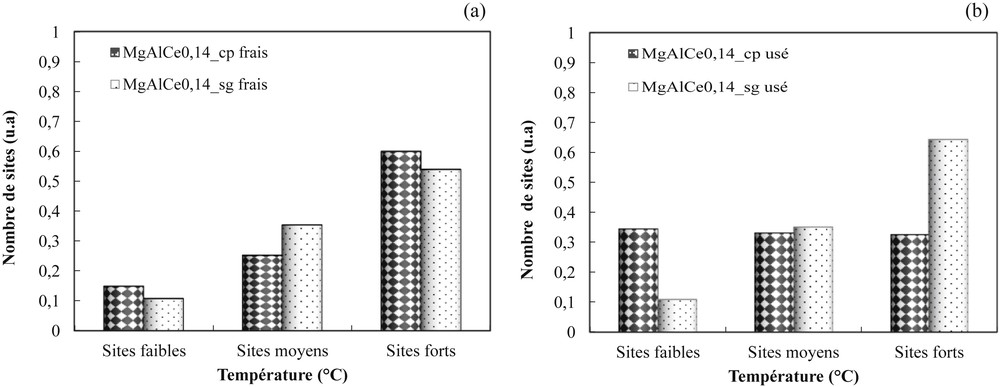
Quantification des sites basiques faibles, moyens et forts des catalyseurs frais et usés MgAlCe0,14 préparés par voie sol–gel et par co-précipitation.
3.2 Test catalytique
Plusieurs cycles de montée en température ont été effectués sur les matériaux afin de vérifier leur stabilité. La Fig. 4 présente les résultats du premier cycle d'oxydation sur les matériaux et les courbes de conversion se décomposent en deux zones distinctes. À basse température (T < 250 °C) des phénomènes d’adsorption importants sont observés sur les catalyseurs contenant MgAl, ce qui explique les conversions non nulles. À plus haute température, l’acide acétique est oxydé essentiellement en CO2. Les phénomènes d'adsorption, limités par la présence de cérium, sont aléatoires de par la complexité des surfaces [30], favorisés à basse température [31], et rendent difficile la comparaison de l'efficacité des différents catalyseurs. Il semble donc intéressant d'effectuer un second cycle catalytique afin de vérifier si ces phénomènes d'adsorption à basse température sont reproductibles ou si une modification de la surface des matériaux a lieu lors du premier cycle. Les courbes de rendement en CO2 présentées sur la Fig. 5 permettent de mieux comprendre les différents événements qui ont lieu au cours de la réaction d'oxydation de l'acide acétique en fonction de la température. En effet, cette figure présente les résultats obtenus lors des deux premiers cycles d'oxydation. La désactivation du catalyseur a été mesurée sur trois cycles catalytiques et aucune variation de l'allure des courbes n'est observée au-delà du second cycle. Dans tous les cas, le catalyseur CeO2 semble le plus actif, puisque c'est pour ce matériau que CO2 est formé à la plus basse température, suivi par MgAlCe0,14 et enfin par MgAl, et ce quelle que soit la méthode de préparation des matériaux.

Conversion de CH3COOH obtenue au cours du 1er cycle d’oxydation de l’acide acétique sur les catalyseurs CeO2, MgAl et MgAlCe0,14 synthétisés par co-précipitation (a) et par voie sol–gel (b).
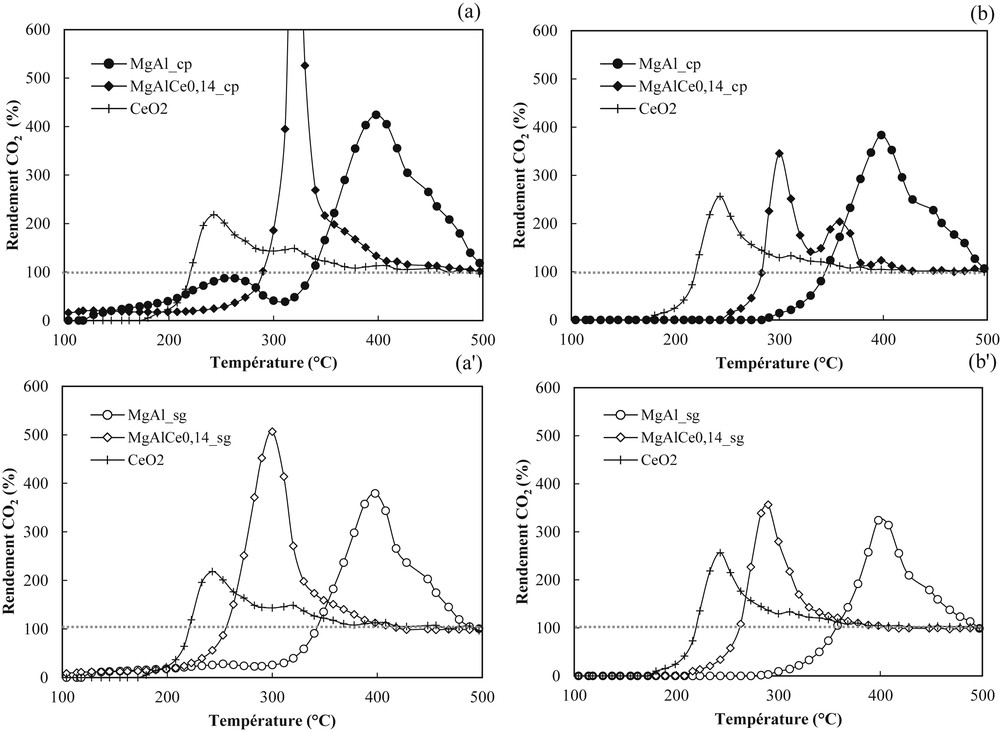
Rendements en CO2 obtenus au cours du 1er et du 2nd cycle d’oxydation de l’acide acétique sur les catalyseurs CeO2, MgAl et MgAlCe0,14 synthétisés par co-précipitation (1er cycle a, 2nd cycle b) et par voie sol–gel (1er cycle a', 2nd cycle b').
Le rendement en CO2 à basse température est quasi-nul, le phénomène d'adsorption de l'acide acétique s'observant sur l’ensemble des matériaux à T < 150 °C. Pour les matériaux MgAl et MgAlCe0,14, l'adsorption et la conversion interviennent simultanément à basse température. L'activité des catalyseurs est faible et le rendement en CO2 entraine la formation d'un un palier en dessous de 20%. Ce palier est d'autant plus marqué que l'adsorption est élevée. À plus haute température, l’activité croît et les rendements en CO2 augmentent au-delà de 100%. Ces rendements, largement supérieurs à 100%, s'expliquent par le fait que l'acide acétique adsorbé et accumulé sur le catalyseur à basse température est totalement désorbé et oxydé lorsque la température augmente. En comparant les échantillons MgAlCe0,14_cp et MgAlCe0,14_sg, l'adsorption est visiblement plus élevée en co-précipitation qu'en sol–gel, puisque la production de CO2 est beaucoup plus importante pour le catalyseur préparé par co-précipitation. Après la fin de la désorption, le rendement en CO2 se stabilise normalement à 100% au-delà de 400 °C. Tous ces phénomènes influencent la réactivité à la surface des catalyseurs. Cependant, la nature et le nombre de sites basiques des solides modifient l’activité des catalyseurs.
3.2.1 Catalyseurs MgAl
En absence de cérium (Fig. 4), une première phase d’activité est observée à 250 °C sur les catalyseurs MgAl_cp et MgAl_sg. Ce pic d’activité, repérable transitoirement par une production de CO2, est beaucoup plus marqué pour MgAl_cp. Lors du premier cycle, à basse température (Fig. 5a et 5b), les rendements maximum en CO2 sont de 87% pour MgAl_cp et de 28% pour MgAl_sg. Au-delà de 300 °C, l'oxydation reprend et devient totale vers 350 °C avec des rendements en CO2 excédentaires (> 100%). Ces résultats sont directement liés à la nature et aux nombres de sites basiques des catalyseurs. En effet, à basse température (100 °C < T ≤ 300 °C), les sites basiques prédominants et actifs sont les sites moyens Mg2+–O2− en dépit de la présence des sites forts O2−. Étant donné que le nombre de sites Mg2+–O2− est plus élevé sur MgAl_cp que sur MgAl_sg, l’activité du matériau co-précipité est plus élevée que celle du catalyseur préparé par voie sol–gel. Par conséquent, le rendement en CO2 est plus élevé sur MgAl_cp. Entre 250 °C et 300 °C, les activités et les rendements en CO2 des catalyseurs diminuent. Ce phénomène peut s’expliquer par la neutralisation des sites actifs basiques moyens (Mg2+–O2−) par les carbonates issus de l’oxydation de l’acide acétique qui a débuté dans cette zone de température. En effet, selon Lavalley [24], les sites basiques O2− favorisent la formation des carbonates via l’interaction acido-basique CO2–O2−. Au cours de la première phase d’activité des catalyseurs, le CO2 formé est probablement chimisorbé sur les sites actifs Mg2+–O2−, lesquels forment des carbonates bidentés et/ou pontés à l’origine de la neutralisation. À plus haute température, les sites forts O2−, prédominants entre 300 °C et 500 °C sont responsables de la conversion totale de l’acide acétique (Fig. 4). Pour confirmer cette hypothèse, un deuxième cycle d’oxydation de l’acide acétique a été effectué sur les mêmes matériaux et les rendements en produits formés ont été comparés au cours des deux cycles réactionnels sur la Fig. 6. D’après les Figs. 6.1 et 6.2, aucun produit n'est formé avant 300 °C lors du second cycle, contrairement à ce qui est observé lors du premier cycle. Cependant, au cycle 2, les secondes phases d’activité des catalyseurs sont similaires à celles du cycle 1, avec des rendements en produits équivalents. Ce résultat confirme notre hypothèse selon laquelle les sites basiques Mg2+–O2− ont été neutralisés au cours du premier cycle d’oxydation de l’acide acétique. L’activité conservée à haute température prouve la bonne stabilité des sites forts O2− à la surface des catalyseurs. Par ailleurs, les Figs. 6.1' et 6.2' montrent que l’activité des sites basiques forts O2− est à l’origine de la formation de produits secondaires tels que l'acétone (CH3COCH3), le formaldéhyde (CH2O) et le méthanol (CH3OH). À partir de la décomposition de l’acide acétique et à l’aide de la littérature [32–36], la Fig. 7 montre le schéma réactionnel proposé afin d’expliquer leurs formations.
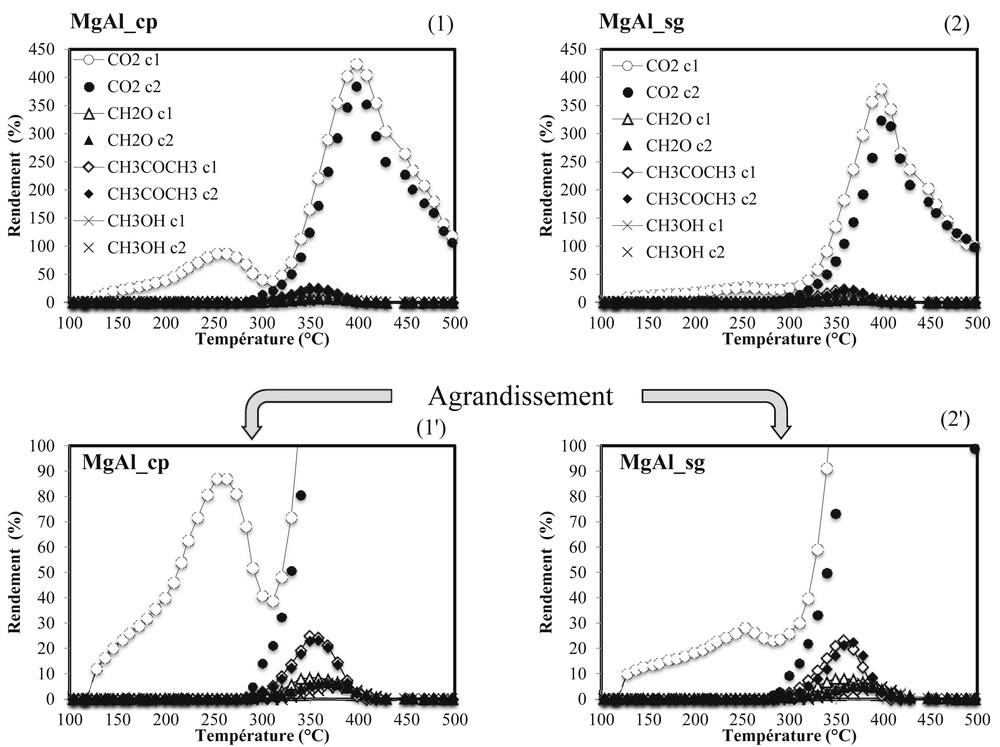
Oxydation de l’acide acétique sur les catalyseurs MgAl_cp (1 et 1') et MgAl_sg (2 et 2'): rendements des produits formés au cours du cycle 1 (c1) [CO2 (), CH2O (), CH3OH (), CH3COCH3 (◊)] et du cycle 2 (c2) [CO2 (●), CH2O (▲), CH3OH (×), CH3COCH3 (♦)].
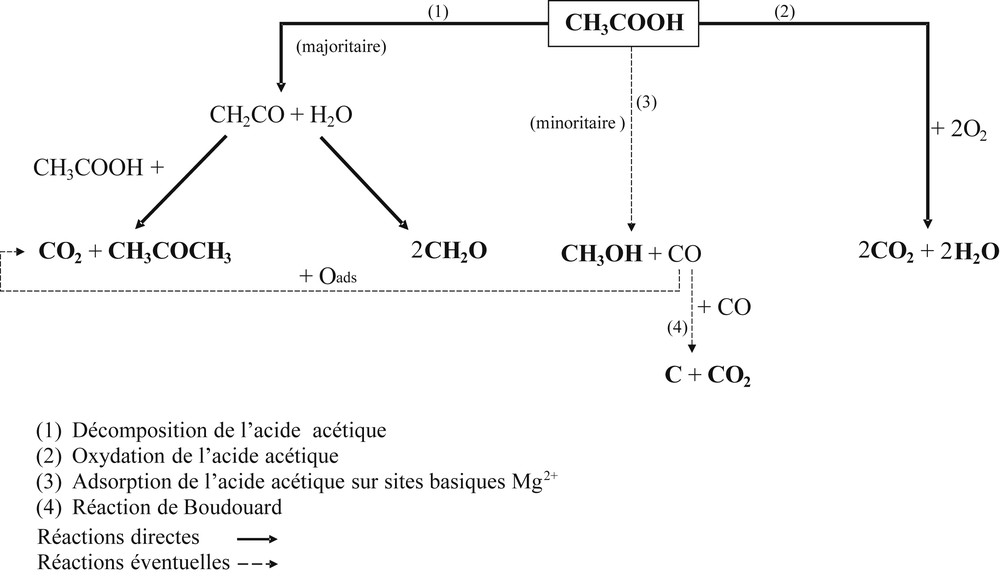
Voies réactionnelles possibles pour la conversion de l’acide acétique.
La décomposition de l’acide acétique adsorbé sur les sites vacants en oxygène génère des réactions parallèles au cours desquelles l’acétone et le dioxyde carbone sont majoritairement formés, par l’intermédiaire d’un cétène (CH2CO) [32,33]. Le formaldéhyde est formé par l’intermédiaire du cétène (sous forme adsorbée) à l’aide des oxygènes de surface du catalyseur [36]. La formation du méthanol est particulière, étant donné qu’elle ne dépend d’aucun produit intermédiaire. Néanmoins, les recherches effectuées par Abate et Kleiber [35] montrent qu’avec suffisamment d’énergie, la dissociation du complexe Mg(CH3COOH), susceptible d’être créé au cours des phases d’adsorption, est facilitée par la rupture des liaisons faibles C–C de l’acide acétique et engendre les espèces réactives CH3CO et OH ou encore CH3ads, COads et OHads adsorbées. Ainsi, il est probable que le méthanol soit formé par la combinaison des espèces CH3ads et OHads. La présence de carbone solide a été suspectée à cause d’un léger noircissement du fritté et des parois du réacteur, après décomposition de l’acide acétique dans un réacteur vide. Ce résultat a conforté notre hypothèse sur la présence de traces de monoxyde de carbone précurseur de coke par la réaction de Boudouard (2CO(g) ⇆ C(s) + CO2(g)). Cette réaction est connue pour être thermodynamiquement totalement déplacée vers la formation de C et CO2 jusqu'à environ 550 °C [37].
3.2.2 Effet de la cérine
Les activités des catalyseurs MgAl et MgAlCe0,14 présentées sur les Fig. (5.A) Et (5.A') ont été comparées au cours du test catalytique. D’une part, à basse température (T < 250 °C), il apparaît que la formation du CO2 est limitée sur le matériau MgAl en présence de CeO2, quelle que soit la méthode de préparation. D’autre part, il semblerait que l’activité des catalyseurs dopés par CeO2 est plus élevée que celle des matériaux MgAl synthétisés par co-précipitation et par voie sol–gel. En effet, le dépôt de cérium à la surface des matériaux imprégnés diminue l’activité des sites moyens Mg2+–O2− et limite la formation du CO2 à basse température. D’après Martin et Duprez [19], la présence de cérine en surface diminue la force de liaison métal-oxygène Mn+–O− des sites basiques des catalyseurs, ce qui réduit finalement la force et le nombres des sites moyens Mg2+–O2− et limite leur activité. Parallèlement à cela, la multiplicité des phénomènes de surface complique la comparaison des catalyseurs entre eux, dans la mesure où la réactivité de surface des espèces présentes fait intervenir, simultanément ou non, la force, la nature et le nombre de sites basiques. En effet, bien que la cérine présente différentes espèces d’oxygène en surface, aucune corrélation évidente avec la force de liaison métal–oxygène Ce–O ou des sites basiques correspondants n’est observée [19]. L’augmentation de la CSO (Tableau 1) confère essentiellement une réductibilité plus élevée, donc une multiplicité de lacunes d’oxygènes favorables à la décomposition de l’acide acétique. En revanche, la basicité est en lien avec le nombre d’espèces métalliques Mg2+ et Al3+ de surface, lesquels font intervenir la force de liaison métal–oxygène. L’activité du solide pourrait être influencée de différentes manières par la réductibilité de la cérine et/ou par la basicité du matériau, étant donné la complexité de la surface.
L'activité des catalyseurs a été évaluée en déterminant les températures pour lesquelles un rendement de 50% en CO2 (TR50) est obtenu au cours de la réaction d’oxydation de l’acide acétique (Tableau 2). Plus la TR50 est basse, plus le matériau est considéré comme actif vis-à-vis de la réaction d'oxydation de l'acide acétique et il est important de noter que la précision des résultats est de ± 1 °C. Le Tableau 2 présente les TR50 pour les différents catalyseurs lors des deux premiers cycles d'oxydation. Dans tous les cas, CeO2 est le matériau le plus actif, avec un TR50 vers 211 °C. Aucune désactivation n'est observée, puisque les courbes des deux cycles sont presque superposables. Les matériaux les moins actifs sont les MgAl, quel que soit leur mode de préparation. Ces matériaux se désactivent légèrement après le premier cycle puisque les TR50 augmentent (+6 à +14 °C pour MgAl_cp et MgAl_sg, respectivement), mais restent stables par la suite (vérifié par un troisième cycle non présenté ici). L'introduction de la cérine sur les catalyseurs a donc un effet positif pour les mélanges d'oxydes qui ont une activité intermédiaire entre celles de la cérine et de MgAl. Lors du premier cycle d'oxydation, MgAlCe0,14_sg est le catalyseur le plus actif par rapport à MgAlCe0,14_cp, puisque la TR50 est mesurée 36 °C plus tôt. L'activité de ces deux matériaux n'est pas modifiée lors du deuxième cycle, bien que le solide préparé par voie sol–gel se désactive légèrement, la TR50 étant à 249 °C au lieu de 239 °C lors du premier cycle. Le catalyseur MgAlCe0,14_cp ne se désactive pas au cours des cycles successifs de réaction. La désactivation observée sur certains matériaux serait due à la neutralisation des sites basiques moyens des catalyseurs au cours du premier cycle. Le catalyseur MgAlCe0,14_sg est plus actif que MgAlCe0,14_cp, alors que les caractérisations effectuées sur les matériaux dopés à la cérine montrent une réductibilité et une basicité plus faible en sol–gel qu’en co-précipitation. Les études menées par Saavedra et al. [38], ont démontré que l’activité des oxydes métalliques est inversement proportionnelle à l’adsorption de carbonates à la surface. En effet, une perte d’activité des sites catalytiques de l’oxyde est observée lorsque le nombre de carbonates en surface est élevé. Il est possible que la formation de carbonates à la surface du catalyseur limite, soit l’adsorption du réactif, soit l’activation de l’oxygène sur les sites actifs. Mais il est également possible que les carbonates se déposent en surface sans réagir ou alors qu'ils participent à la réaction en tant qu’intermédiaires réactionnels. Dans tous les cas, la conséquence observée est une baisse d'activité spécifique. Par conséquent, l'activité du catalyseur MgAlCe0,14_cp pourrait être limitée par sa forte basicité.
Températures pour lesquelles un rendement de 50% en CO2 est obtenu lors de la réaction d'oxydation de l'acide acétique, pour les deux premiers cycles d'expériences.
| Échantillons | TR50 (°C) | |
| Cycle 1 | Cycle 2 | |
| MgAl_cp | 322 | 328 |
| MgAl_sg | 325 | 339 |
| MgAlCe0,14_cp | 275 | 276 |
| MgAlCe0,14_sg | 239 | 249 |
| CeO2 | 212 | 211 |
4 Conclusion
Les phénomènes d’adsorption sont largement présents à basse température sur les catalyseurs préparés et favorisés par la nature basique des matériaux. L’ajout de cérium améliore l’activité des catalyseurs préparés par co-précipitation et sol–gel. D’une part, la capacité de stockage en oxygène augmente considérablement à cause de la présence de lacunes d’oxygène en surface issues du cérium et, d’autre part, la basicité des échantillons est améliorée grâce au caractère basique de la cérine. Cependant, au cours de l’oxydation catalytique, la complexité des surfaces fait intervenir le caractère basique et la réductibilité des catalyseurs. La basicité augmente l’interaction acido-basique à la surface des catalyseurs et la réductibilité augmente l’activité des matériaux. Une très forte basicité engendre la formation de carbonates à la surface, lesquels limitent la disponibilité des sites actifs et affectent l'activité des catalyseurs. L'étude de ces matériaux est donc complexe et il n'est pas possible de trouver le paramètre déterminant afin de favoriser l'activité. Des études plus poussées comparant une série de catalyseurs avec une quantité de cérium croissante pourraient permettre de mieux cerner les mécanismes d'oxydo-réduction impliqués dans cette réaction d'oxydation. Une modification de la basicité peut également être envisagée afin de mettre en lumière la participation du caractère basique des catalyseurs à la réaction.