

1 Introduction
In recent years, nanotube materials have been intensely investigated due to their physical and chemical properties [1–9]. It is well known that these properties are strongly dependent on structural organization and also on the presence of vacancies and impurities [7,10–15]. Gallium nitride is an important material due to its optoelectronic properties and high thermal and mechanical stability, which is also appropriate to produce light-emitting diodes (LEDs) with short wavelengths [13,16–23]. Wurtzite is the thermodynamically stable phase of GaN, which is a semiconductor material and shows a band gap close to 3.4 eV [18,24–26].
GaN nanotube materials have attracted wide theoretical and experimental interest [20,27–33]. Hemmingsson et al. observed a band gap of 3.46 and 3.75 eV for GaNNTs [27]. Yang et al., using DFT, found a band gap of 1.72 eV for zigzag GaNNTs with 5.35 Å diameter [30]. They also showed that the band gap increases with the diameter size. The Ga 3d band was found at 19.9 eV of binding energy from PES spectra with a band gap of 3.37 eV [28]. However, the energy gap of small nanotubes is not well known, because this depends on structural organization [31]. Consequently, the study of nanotube geometry is important to better understand their electronic and magnetic properties [11,34–37].
Many methodologies have been used for building inorganic nanotube models, based on graphene [20,35,38] or on the crystalline structure [39,40]. In this work, we used cluster models to generate gallium nitride crystal coordinates in the wurtzite phase for the (0001) surface to obtain nanotubes in the armchair and zigzag conformations. The (0001) surface of GaN was used as it is the most studied termination [41–43].
1.1 Methodology
The lattice parameters of the GaN crystal in the wurtzite phase [44] were used to generate the coordinates of the (0001) surface, and the sheet was wrapped to form the nanotube (Fig. 1).
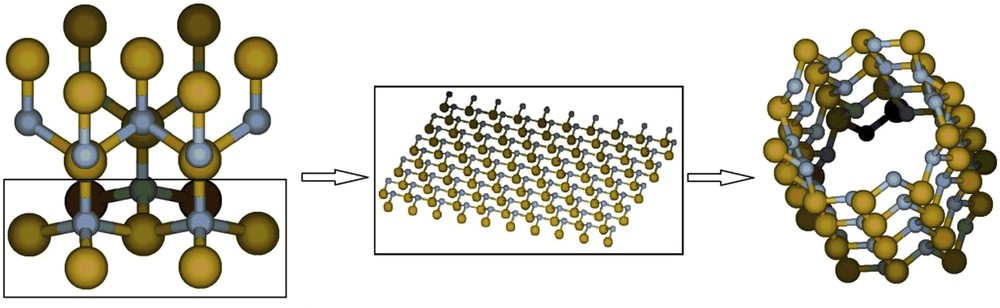
GaN cell, (0001) surface and modeled nanotube. Large spheres represent Ga atoms, while small spheres represent N atoms.
The nanotube geometry was fully optimized using the PM7 semiempirical method [45] as implemented on the MOPAC 2012 package [46]. PM7 shows small average errors compared to other semiempirical parametrizations [46]. The PM7 method was also used to calculate strain energy (Es), energy variation (ΔE), charge distribution, dipole moment and orbital contribution.
| (1) |
| (2) |
The optimized coordinates were used as the input for the single-point Hartree–Fock (HF) and B3LYP methods with the 6-311G basis set, to calculate the strain energy (Eq. (1)), energy variation (Eq. (2)), charge distribution, dipole moment and the frontier orbitals, and the difference between the highest occupied molecular orbital and lowest unoccupied molecular orbital (|HOMO–LUMO|). First principles calculations were carried out using Gaussian 03 software [47].
2 Results and discussions
The strain energy (Es) values are shown in Table 1. As expected, the analysis of nanotube stability, comparing the strain energy of nanotubes in both conformations, indicates that the nanotube structures are more stable than the isolated surface. The variation of energy due to the nanotube growth along the z-direction parallel to the surface (Fig. 2) has shown that the growth is directly proportional to the nanotube stability. The GaNNTs obtained experimentally indicated a length close to 1 μm [12].
Strain energy (eV) for armchair (5,5)10 and zigzag (10,0)10 models.
| Armchair | Zigzag | |
| PM7 | −1.856 | −1.732 |
| HF/6-311G | −1.767 | −1.578 |
| B3LYP/6-311G | −1.695 | −1.619 |
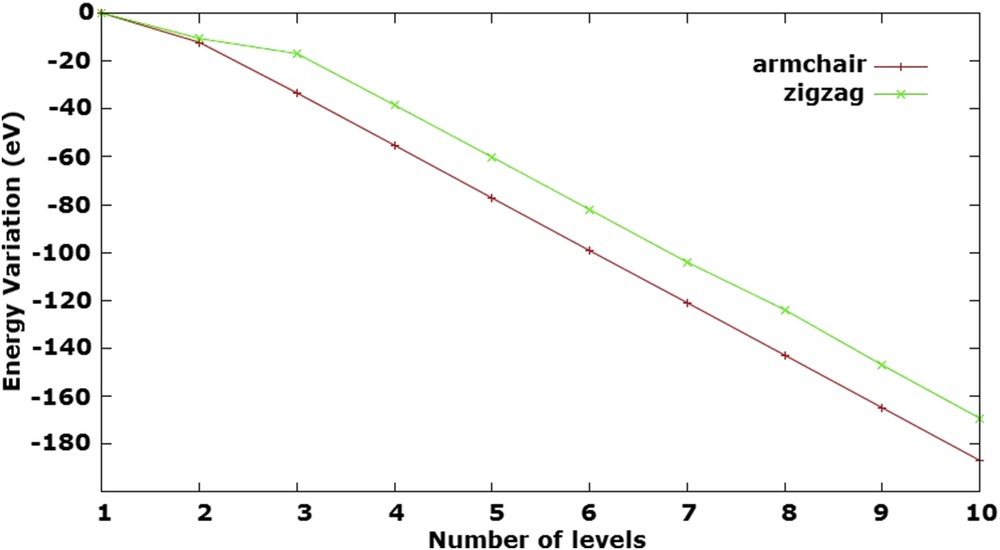
Energy variation related to the nanotube growth for armchair (5,5) and zigzag (10,0) models.
Comparing the strain energy, we found a difference of −0.076 eV for the GaN unit (B3LYP/6-311G) between armchair and zigzag conformations. The armchair was found as the most stable conformation. However, it is possible to note the distortion in the extremities of this armchair conformation (Fig. 3). In order to balance the effects of free valences (dangling bonds) for unsaturated models, the structures were saturated with hydrogen. The results show no distortion for the optimized saturated structures (Fig. 2c). It is noteworthy that these models were saturated in order to control the border effects.
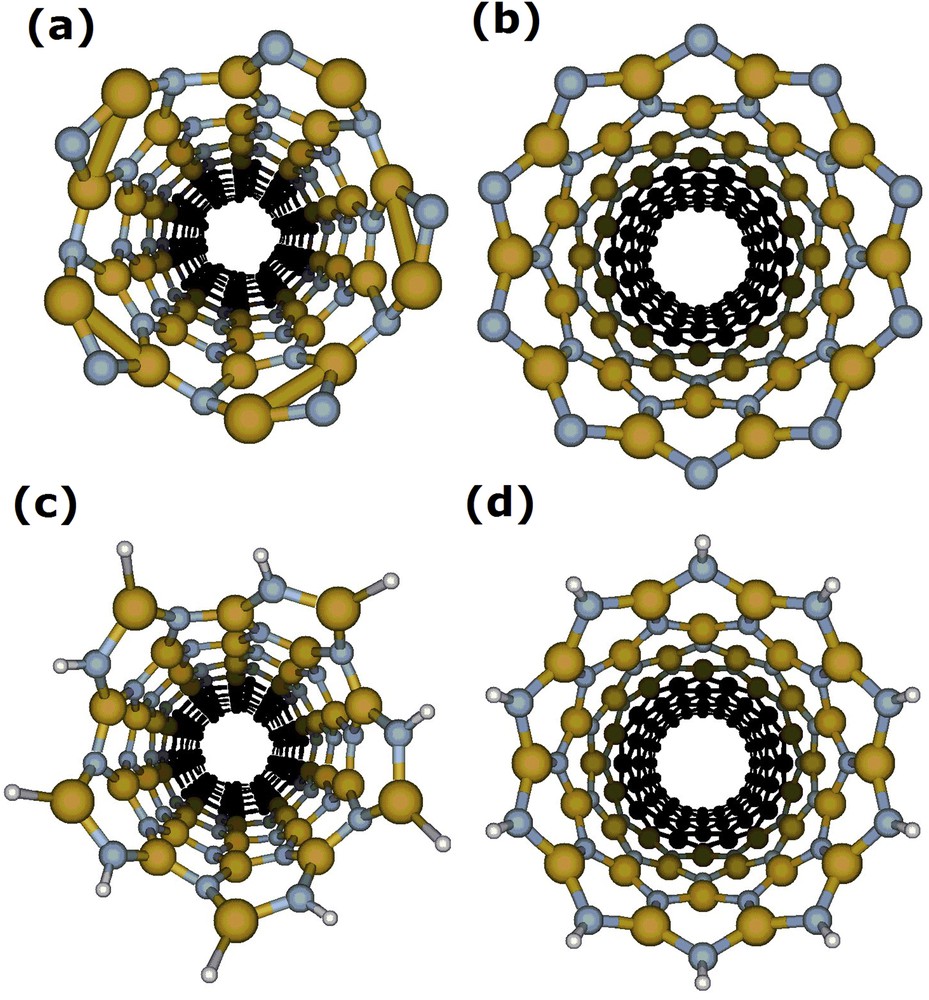
(A) armchair (5,5) and (B) zigzag (10,0) unsaturated GaNNT models and (C) armchair (5,5) and (D) zigzag (10,0) saturated GaNNT models after optimization. Large spheres represent Ga atoms and small spheres represent N atoms.
Table 2 shows the semiempirical results for the bond length, diameter and nanotube length, as well as the dipole moment after optimization. HF and DFT data are single-point results. It is possible to note that the models present two different values for the radius. This is due to the differing atomic position between the Ga and N atoms. It was expected to obtain the increase of the inner radius due to the repulsive forces acting on the N atoms. The external diameter for the armchair structure is in agreement with that obtained by Beigi [33] and Moradian et al. [31] in their study regarding the structural and electronic properties of single-walled GaNNTs. The armchair models have the largest length due to the structure of this nanotube model.
Structural parameters and dipole moment of GaNNT models in armchair (5,5)10 and zigzag (10,0)10.
| Length/Å | Diameter/Å | Dipole/Debye | |||||||||
| GaN bond | Nanotube | Outer radius | Inner radius | PM7 | HF/6-311G | B3LYP/6-311G | |||||
| Armchair (unsaturated) | 1.84 | 30.45 | 8.77 | 8.13 | 0.023 | 0.0200 | 0.0169 | ||||
| X = 0.01 | Y = 0.02 | Z = −0.01 | X = 0.01 | Y = −0.01 | Z = 0.00 | X = 0.01 | Y = −0.01 | Z = 0.00 | |||
| Armchair (saturated) | 1.84 | 32.65 | 8.94 | 8.77 | 0.014 | 0.0202 | 0.0133 | ||||
| X = 0.01 | Y = 0.02 | Z = −0.00 | X = 0.01 | Y = −0.02 | Z = 0.00 | X = 0.01 | Y = −0.01 | Z = 0.00 | |||
| Zigzag (unsaturated) | 1.84 | 25.78 | 9.46 | 8.80 | 43.235 | 0.2110 | 7.2875 | ||||
| X = −0.00 | Y = 0.02 | Z = −43.2 | X = −0.21 | Y = 0.01 | Z = 0.01 | X = 7.22 | Y = 0.22 | Z = −0.99 | |||
| Zigzag (saturated) | 1.84 | 28.00 | 10.30 | 10.13 | 17.708 | 7.1231 | 7.5616 | ||||
| X = −0.00 | Y = 0.01 | Z = −17.7 | X = 7.12 | Y = −0.01 | Z = −0.00 | X = 7.56 | Y = −0.01 | Z = −0.00 |
For the saturated models, we observed a larger nanotube length and diameter compared to those of the unsaturated models. The increase in length was due to the hydrogen atoms in the extremities. Moreover, the variations found in diameter are probably related to saturation effects. The saturated models are more similar to the structure achieved on carbon nanotubes. When we take into consideration the difference between the inner radius and outer radius, the value is smaller for the saturated models.
On analyzing the dipole moment (Table 2), we observed that the armchair structure has a dipole moment closer to zero, suggesting a non-polar structure. Otherwise, the zigzag conformation indicates a small dipole moment using ab initio methods and a high dipole moment using the PM7 method. This trend suggests polar characteristics for the zigzag conformation. The results obtained by the DFT method show that the armchair models are non-polar, whereas the zigzag models have a dipole moment ranging from 7 to 8 Debye in the x-axis direction.
The sum of Mulliken charge by layer (Table 3) confirms the non-polar characteristic for the armchair conformation, due to the way in which the charges are distributed for this nanotube. On the other hand, the zigzag conformation shows that the total charges are different for every extremity, suggesting a polar structure in accordance with the dipole moment.
Sum of the Mulliken atomic charges by layer for unsaturated and saturated models.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | ||
| Armchair (unsaturated) | PM7 | 0.37 | −0.39 | −0.02 | 0.05 | −0.01 | −0.01 | 0.05 | −0.02 | −0.39 | 0.37 |
| HF/6-311G | 0.68 | 0.03 | −0.32 | −0.15 | −0.23 | −0.23 | −0.15 | −0.32 | 0.03 | 0.68 | |
| B3LYP/6-311G | 0.71 | 0.14 | −0.34 | −0.22 | −0.28 | −0.28 | −0.22 | −0.34 | 0.14 | 0.71 | |
| Armchair (saturated) | PM7 | 0.17 | −0.11 | −0.01 | 0.00 | −0.05 | −0.05 | 0.00 | −0.01 | −0.11 | 1.69 |
| HF/6-311G | 0.80 | −0.03 | −0.34 | −0.15 | −0.28 | −0.28 | −0.15 | −0.34 | −0.03 | 0.56 | |
| B3LYP/6-311G | 0.87 | 0.05 | −0.32 | −0.32 | −0.25 | −0.32 | −0.25 | −0.34 | 0.05 | 0.81 | |
| Zigzag (unsaturated) | PM7 | −1.89 | −0.70 | 0.09 | −0.02 | −0.05 | −0.03 | 0.01 | 0.02 | −0.27 | 2.82 |
| HF/6-311G | −4.02 | −0.80 | −0.24 | −0.35 | −0.22 | −0.18 | −0.23 | −0.27 | 0.23 | 6.07 | |
| B3LYP/6-311G | −2.90 | −0.58 | −0.31 | −0.45 | −0.29 | −0.25 | −0.32 | −0.34 | 0.59 | 4.86 | |
| Zigzag (saturated) | PM7 | −2.40 | −0.26 | 0.21 | −0.07 | −0.06 | −0.02 | −0.03 | −0.03 | 0.03 | 2.56 |
| HF/6-311G | −4.72 | −0.10 | −0.12 | −0.46 | −0.20 | −0.20 | −0.24 | −0.29 | −0.10 | 3.35 | |
| B3LYP/6-311G | −3.38 | 0.00 | −0.16 | −0.55 | −0.29 | −0.28 | −0.31 | −0.35 | 0.00 | 3.20 |
We also calculated the gap (HOMO-LUMO) values using the HF and B3LYP with the 6-311G basis set, for saturated and unsaturated models (Table 4). It is possible to note the difference between the energy gap for unsaturated models, while saturated models show accordance between calculated gap values. We observed that the HF method indicates larger values of gap for all models. This trend of the HF method is expected. For HF calculations, only the unsaturated zigzag conformation showed semiconductor characteristics, whereas the DFT calculations showed semiconductor characteristics for all models. Hemmingsson et al. [27] observed two peaks for GaNNTs measured from the low-temperature time-resolved photoluminescence spectrum at 3.47 eV and 3.75 eV. The gap values obtained from the saturated models using the density functional theory are close to the value of the experimental second peak (3.75 eV [27]) for both models (Table 4).
Energy gap (eV) obtained by ab initio methods of GaNNT models for armchair (5,5)10 and zigzag (10,0)10 conformations.
| Armchair | Zigzag | |||
| Unsaturated | Saturated | Unsaturated | Saturated | |
| HF/6-311G | 8.512 | 9.213 | 2.793 | 9.367 |
| B3LYP/6-311G | 2.937 | 3.820 | 0.108 | 3.922 |
The density of states for saturated and unsaturated models is depicted in Fig. 4. It was found that after saturation the models showed similar densities of states. We concluded that although the border effects from unsaturated models do not substantially alter the polarity of structures, there was a major influence on the gap of these nanotubes.
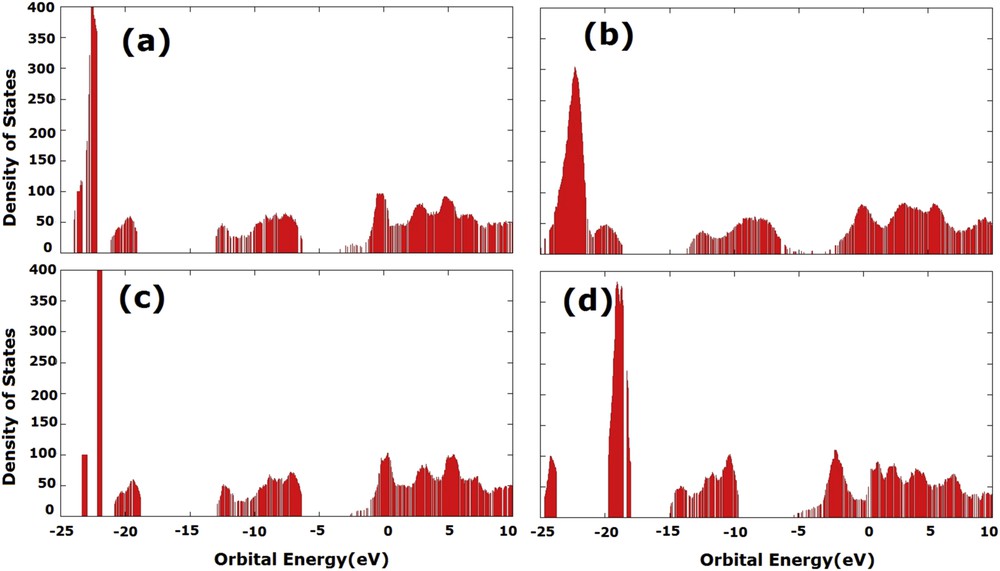
Density of states at the B3LYP level: unsaturated (a) armchair and (b) zigzag; saturated (c) armchair and (d) zigzag.
The gap value and DOS obtained with the cluster model at the B3LYP level is in good agreement with results obtained by Srivastava et al. [11], using the Density Functional Theory (DFT) with Linear Combination of Atomic Orbital (LCAO), and Lee et al. [18], using a Self-Consistent Charge Density Functional based Tight Binding method (SCC-DFTB).
Figs. 5 and 6 show the molecular electronic potential maps HOMO and LUMO for armchair and zigzag conformation, respectively. It is easily seen that the HOMO is distributed on one of the edges for all models. However, the same does not occur with the LUMO. In the armchair models, the LUMO is mainly distributed in the atoms in the middle of the nanotube for the saturated and unsaturated models. For unsaturated models, in the zigzag conformation, the LUMO is composed by nitrogen atoms on the border. However, after the saturation of the model, the LUMO orbital contribution was dislocated to atoms at the middle of the nanotube.

Saturated model results of MEP (a), HOMO (b) and LUMO (c), and unsaturated model results of MEP (d), HOMO (e) and LUMO (f) for armchair (5,5)10.

Saturated model results of MEP (a), HOMO (b) and LUMO (c), and unsaturated model results of MEP (d), HOMO (e) and LUMO (f) for zigzag (10,0)10.
3 Conclusions
We have studied GaN single-walled nanotubes using PM7, RHF and DFT methods. We have found that the armchair model is the most stable due to homogeneous charge distribution by layer. The distribution of charges is different for each conformation, influencing the polarity of the nanotube. For the armchair conformation, saturation of the model presents no influence on the density of states, while the zigzag conformation shows different behavior between saturated and unsaturated models. This is probably due to the polarization found in the zigzag model shown by the dipole moment and layers charge.
Another important result is regarding the difference between the HOMO and LUMO in relation to the nanotube conformation. In the zigzag and armchair conformations, the HOMO is mainly distributed over the border atoms, while the LUMO is located in the middle of the armchair conformation. We studied the border effects by adding saturation on the nanotube structures. It was observed that when using saturation the LUMO was distributed mainly in the middle levels for both conformations. This fact explains the similarity between the DOS of armchair (saturated and unsaturated) and zigzag (saturated) models.
It was also noted that saturated models show similar gap values using methods based on Hartree–Fock (RHF) and Density Functional (B3LYP) theories for armchair and zigzag conformations. Our results support the importance of structural organization in the electronic structure and properties.
Acknowledgements
The authors are grateful for the financial support from CNPq (306945/2015-0) and CAPES.