

1 Introduction
Hydrosilylation is a powerful tool to functionalize organic molecules, and it represents one of the most important routes in obtaining organosilicon compounds [1]. The addition of SiH bonds to unsaturated compounds could be carried out by free radical chain reactions or more traditionally by the use of different catalysts such as platinum or other transition metals. For this reaction, the Karstedt catalyst of the empirical formula [Pt2(CH2CHSi(Me)2OSi(Me)2CHCH2)3], which follows a mechanism involving colloids, has predominated in recent years [2]. Recent discoveries in a broad range of applications such as electrical, magnetic, optical, biomedical, coatings, aerospace, and catalysis greatly increased the interest in incorporation of transition metals into organic monomers and polymers [3,4].
Since its discovery in 1951, ferrocene has quickly attracted the attention of the scientific and technical community on account of its interesting chemistry [5–7]. Researchers started to develop synthetic strategies leading to ferrocene derivatives and explored their applications in a wide range of scientific areas [8]. Due to the favorable electronic properties of ferrocene and its ease of functionalization, these compounds have found many applications in materials science, including sensors, [9–15] catalysts, [16–19] electroactive materials [20–22] and aerospace materials.
Ferrocene derivatives are a well-known class of one-electron donors which exhibit well established reversible redox couples. As a consequence, ferrocene derivatives, particularly those possessing functionalized tethers, have emerged as strong candidates for molecular electronic devices, electro-optical materials, multielectron redox catalysts and electrode surface modifiers [23,24]. There are several methods known for the formation of vinyl or propenylferrocene. Derivatives with longer alkenyl chains are hardly known in the literature. Previous methods for the synthesis of alkenylferrocenes were based on the dehydration and dehydrohalogenation reactions of the corresponding ferrocenylalkanols and ferrocenylalkylhalides with a suitable reagent, respectively [25,26].
Based on our interest in ferrocenyl chemistry here, we report the synthesis of binuclear ferrocenyl compounds using the hydrosilylation strategy. Besides the electrochemical properties of the synthesized ferrocenyl compounds are reported in the present article.
2 Result and discussion
Alkylferrocenes (3a–c) were synthesized by acylation of ferrocene with acyl chlorides in dry CH2Cl2 in the presence of AlCl3 as the catalyst. The reaction was subsequently followed by the reduction of carbonyl compounds (2a–c) by adding NaBH4 in diglyme to produce the desired compounds in good yields (Scheme 1).

Synthesis of 4-chlorobutylalkylferrocenes (5a–d).
4-Chlorobutylferrocene (5a) was synthesized by the acylation reaction as reported in the literature [27,28]. Acylation of alkylferrocenes (3a–c) with 4-chlorobutanoyl chloride in dry CH2Cl2 and AlCl3 as the catalyst which subsequently continues by in situ reduction of carbonyl compounds (4b–d) in the presence of NaBH4 in diglyme, produces 4-chlorobutylalkylferrocenes (5b–d) in high yields (Table 1 and Scheme 1).
Synthesis of 4-chlorobutylalkylferrocenes (5a–d).
| Entry | Product | Yield% |
| 1 | 5a | 90 |
| 2 | 5b | 89 |
| 3 | 5c | 88 |
| 4 | 5d | 88 |
3-Butenylferrocene (6a) and 3-Butenylalkylferrocenes (6b–d) were synthesized with the elimination reaction of 4-chlorobutylferocene (5a) and 4-chlorobutylalkylferocene derivatives (5b–d) by sodium tert-butoxide in DMSO at 60 °C in good yields, respectively [26]. This reaction also was carried out by simple bases such as KOH or NaOH in diglyme and DMSO, but the rate of conversion was less than when sodium tert-butoxide was used (Scheme 2). In 48 h, compound 6a was obtained in 80% yield using KOH at 80 °C while at 60 °C the output decreases to 65% yield in the same reaction time (Table 2, Entry 3).
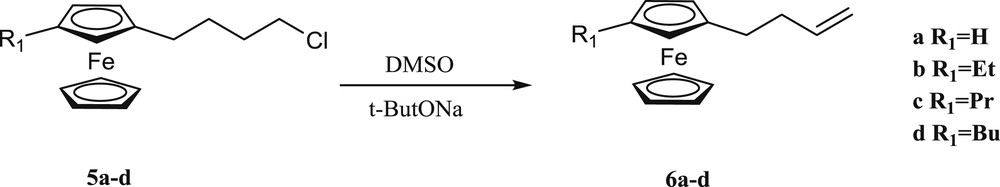
Synthesis of (3-butenyl)alkylferrocenes.
Synthesis of 3-butenylferrocene (6a).
| Entry | Temperature °C | Solvent | Base | Time/h | Yield/% |
| 1 | 60 | Diglyme | NaOH | 36 | 5 |
| 2 | 60 | DMSO | NaOH | 36 | 10 |
| 3 | 60 | DMSO | KOH | 48 | 65 |
| 4 | 80 | DMSO | KOH | 48 | 80 |
| 5 | 60 | DMSO | t-BuONa | 36 | 88 |
Similarly, the reaction in diglyme was not successful and only 5% product was obtained (Table 2, Entry 1). By NaOH in DMSO, 3-butenylferrocene was obtained in 10% yield after 36 h (Table 2, Entry 2).
(4-Ferrocenylbutyl)dimethylsilane derivatives were synthesized by the Grignard reaction between 4-chlorobutylferrocene (5a) [29], (4-chlorobutyl)ethylferrocene (5b) and chlorodimethylsilane in THF in 89 and 91% yields, respectively (Scheme 3) [30,31].
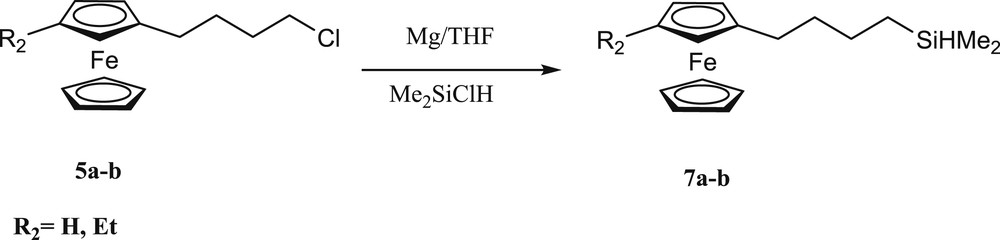
Synthesis of (4-ferrocenylbutyl)dimethylsilane derivatives.
For the synthesis of binuclear ferrocenyl based organosilane compounds initially we treated the 3-(butenylferrocene) (6a) with (4-ferrocenylbutyl)dimethylsilane (7a) in dry hexane with the Karstedt catalyst at room temperature. After 3 days the desired product was obtained in low yield (Table 3, Entry 1); in this case (4-ferrocenylbutyl)dimethylsiloxane was formed as the main product under reflux conditions. Using THF gave the desired bis(4-ferrocenylbutyl)dimethylsilane (8a) in 10% yield.
Synthesis of bis(ferrocenylbutyl)dimethylsilane (8a).
| Entry | Compound | Compound | Solvent | Time | Yield% |
| 1 | 6a | 7a | Hexane | 3 day | 20 |
| 2 | 6a | 7a | THF | 36 h | 10 |
| 3 | 6a | 7a | Toluene | 36 h | 91 |
Finally in dry toluene at room temperature after 24 h the product (8a) was obtained with 91% yield (Table 3, Entry 3). The novel binuclear ferrocenyl based organosilane compounds 8a–g were synthesized according to this procedure in 80–91% yields (Table 4). The results showed that alkyl substituted ferrocenyl moieties are less reactive than others present in this reaction. 1H NMR spectra of the obtained compound showed that the only formed product was alpha and no beta type was observed (Scheme 4).
Bis[4-(alkylferrocenyl)butyl]dimethylsilane derivatives (8a–8g).
| Entry | Butenylferrocenes | Silane | Product | Yield%∗ |
| 1 | 6a | 7a | 8a | 91 |
| 2 | 6b | 7a | 8b | 91 |
| 3 | 6c | 7a | 8c | 87 |
| 4 | 6d | 7a | 8d | 81 |
| 5 | 6a | 7b | 8b | 91 |
| 6 | 6b | 7b | 8e | 83 |
| 7 | 6c | 7b | 8f | 86 |
| 8 | 6d | 7b | 8g | 80 |
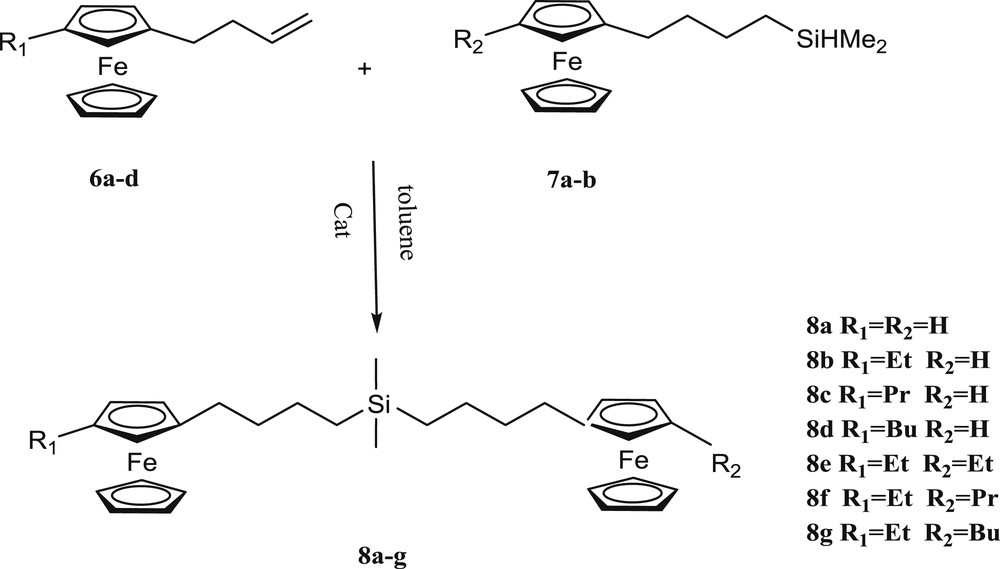
Synthesis of bis[4-(alkylferrocenyl)butyl]dimethylsilane derivatives.
Symmetrical binuclear ferrocenyl based organosilane compounds 8a and 8e were also synthesized by the Grignard reaction between 5a and 5b with dimethyldichlorosilane in THF under reflux conditions in 92 and 84% yields, respectively (Scheme 5).

Synthesis of a symmetrical binuclear ferrocenyl based organosilane by the Grignard reaction.
3 Cyclic voltammetry
The electrochemical behavior of the synthesized compounds has been studied by cyclic voltammetry at different scan rates (mV/s) 25, 50, 100, 150, 200 and 250 in CH3CN/0.100 M LiClO4. The resulting diagrams represent pseudo-reversible to totally reversible behavior which can be attributed to the single-electron oxidation–reduction reaction of the ferrocene rings. In the diagrams plotted for each of the synthesized compounds, it can be observed that the anodic and cathodic peaks do not change with the potential scan rate. The difference between anodic and cathodic peak potentials is about 75 mV, which is expressed as a single-electron reversible reaction. Plotting of the anodic and cathodic peak currents versus square root of the scan rate represents a straight line with a regression coefficient of 0.99, confirming that redox reactions in the system are diffusion-controlled kinetically fast reactions. In other words, the electron transfer process is rapid and oxidation–reduction half-reactions take place at high speeds. CV voltammograms of compounds (8a) are given in Fig. 1.
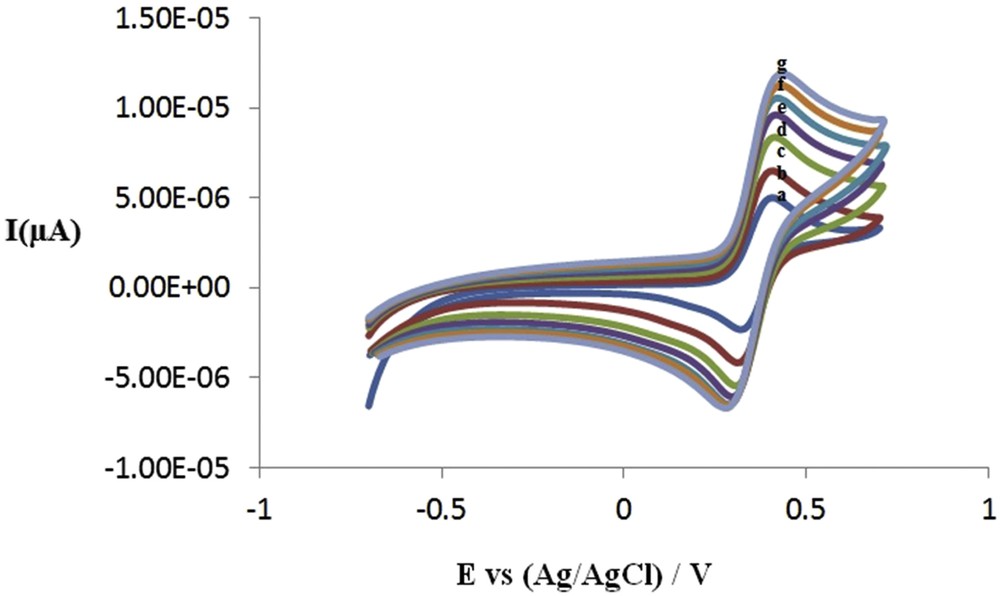
Cyclic voltammetry of bis(butylferrocen)dimethylsilane (8a) in CH3CN/LiClO4. (a) 0.025 (b) 0.05 (c) 0.1 (d) 0.15 (e) 0.2 (f) 0.25 (g) 0.30 V s−1.
In order to compare the influence of the substituents on the redox ability of Fe (II), we carried out electrochemical studies on the synthesized ferrocene derivatives (5a–5d), (6a–6d) and (8a–8g). CV experiments performed in dry CH3CN/0.100 M LiClO4 exhibited reversible voltammetric behavior for the ferrocenyl group in this compounds with ΔEp = Epa − Epc ≤ 0.08 V at scan rates up to 0.25 V s−1 (Table 5). Cathodic and anodic peak current ratios measured for the derivatives were in the range of 1.02 < ipc/ipa < 1.15, and Ep values were independent of the scan rate.
Potentials (V) and current (μA) data for 1.0 mM solutions of the ferrocenyl containing compounds in CH3CN/0.100 M LiClO4 at 25 °C.
| Compound | ΔEp | ipc/ipa | Compound | ΔEp | ipc/ipa |
| 5a | 0.071 | 1.04 | 8a | 0.065 | 1.12 |
| 5b | 0.076 | 1.04 | 8b | 0.070 | 1.10 |
| 5c | 0.072 | 1.02 | 8c | 0.063 | 1.08 |
| 5d | 0.073 | 1.00 | 8d | 0.069 | 1.11 |
| 6a | 0.060 | 1.07 | 8e | 0.060 | 1.14 |
| 6b | 0.061 | 1.09 | 8f | 0.061 | 1.15 |
| 6c | 0.065 | 1.11 | 8g | 0.068 | 1.13 |
| 6d | 0.063 | 1.12 |
All the compounds exhibit only one pair of well-defined redox peak in CH3CN indicating the existence of only one kind of electroactive center in their structure which corresponds to their ferrocenyl group. Fig. 3 shows an example of typical voltammograms of (4-chlorobutyl)ferrocene in CH3CN/0.100 M LiClO4. The influence of the scan rate on the voltammetric response of the ferrocenyl compounds in the range of 0.025–0.25 V s−1 was investigated. As seen, the anodic and cathodic peak currents increased, meanwhile the elevation in scan rate, the plots of the anodic and cathodic currents versus the square root of scan rates (v1/2) show a linear relationship (Fig. 2).
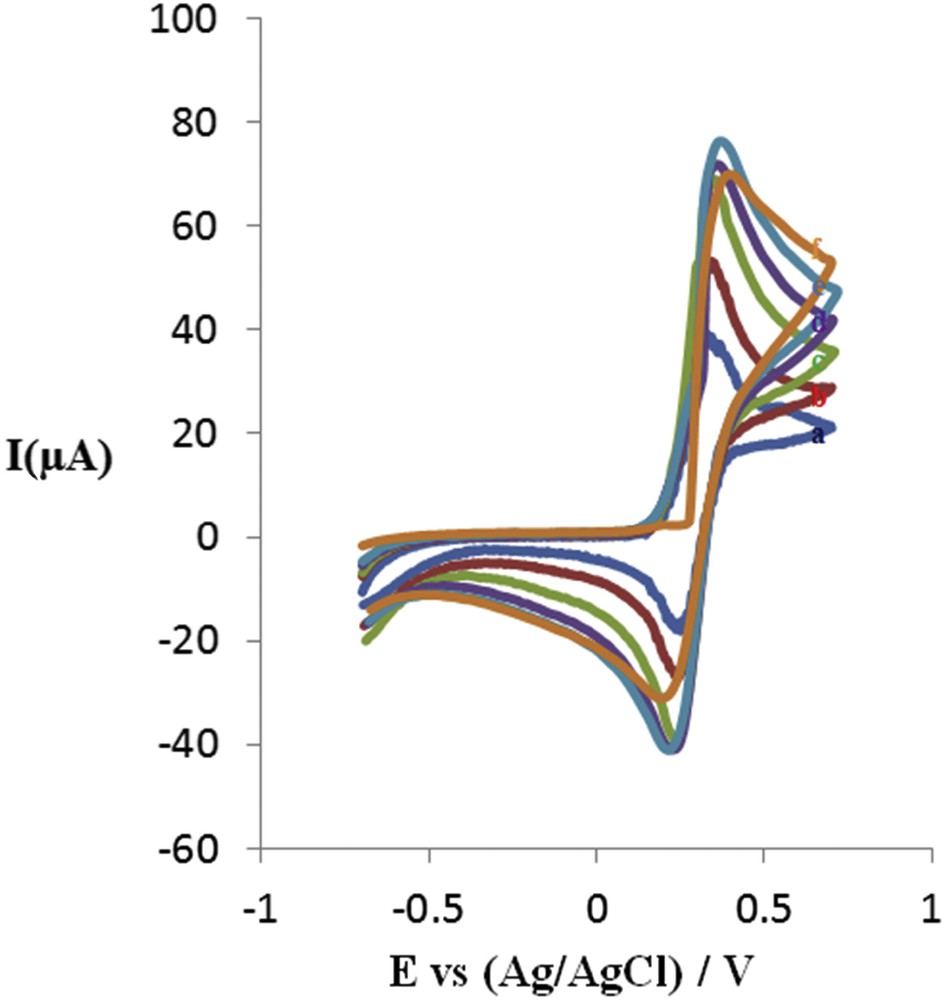
CV curves of the (4-chlorobutyl)ferrocene in different scan rates. 0.025 0.05 0.1 0.15 0.2 0.25 V s−1.

Linear relationship between the A) cathodic peak current, B) anodic peak current and the square root of scan rates.
Both reduction and oxidative peak currents increase obviously as the Fc unit increases in the structure of compounds (Fig. 4). The elevation of the Fc unit is responsible for the increase of electroactive substances; the charge generated in the reaction became a factor leading to a gradual increase in the peak currents as it forms during the reaction. These compounds can be potentially used as new electroactive materials for sensors and other electrochemical systems due to their well reversible redox behavior and the ability to tailor functional groups without affecting the electrochemical aspects.
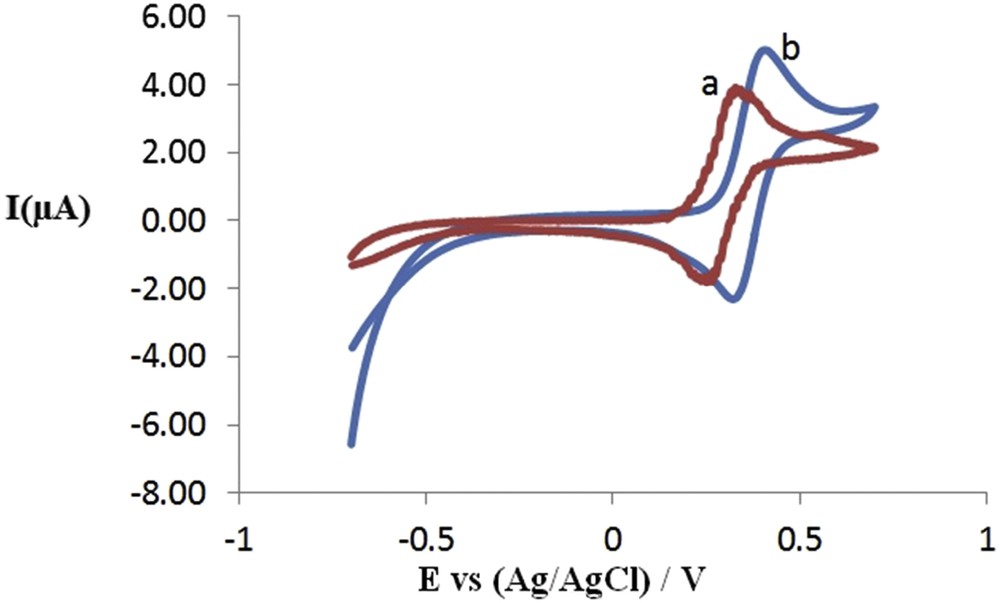
CV curves for 1.0 mM of 4-chlorobutylferrocene bis(4-ferrocenylbutyl)dimethylsilane in CH3CN/0.100 M LiClO4 at 250 mV s−1.
4 Conclusion
In summary, we have reported the design and synthesis of some binuclear ferrocenyl based organosilane compounds by the reaction of (4-ferrocenylbutyl)dimethylsilane or [4-(ethylferrocenyl)butyl]dimethylsilane with alkenylferrocene derivatives, under the Karstedt catalyst at room temperature. Meanwhile we show a simple and convenient method for the preparation of 3-butenylalkylferrocenes from 4-chlorobutylalkylferrocenes under mild conditions. The electrochemical behavior of binuclear ferrocenyl based organosilane compounds also was investigated. As the result the relationship between the peak currents and the square root of the scan rate showed that the electrode processes were diffusion controlled.
5 Experimental
5.1 Solvents and reagents
All chemicals were either prepared in our laboratory or purchased from Merck, Fluka, Sigma, Aldrich and Yantai Suny Chem. International Co., Ltd. The commercial solid reagents were used without further purification. Liquid reactants were distilled prior to use. Solvents were dried and distilled prior to use according to standard laboratory practices; water was double distilled. Toluene and THF were dried by refluxing under an argon atmosphere over sodium wire and distilled directly before use. Column chromatography was performed on silica gel 60 (Merck, grain size 0.063–0.2 mm) and n-hexane was used as the eluent. All reactions were carried out under an atmosphere of argon in oven-dried glassware while they were magnetically stirred.
5.2 Instrumentation
The 1H and 13C NMR spectra were recorded with a Bruker FT-400 MHz spectrometer at room temperature using CDCl3 as the solvent, while the chemical shifts are presented as delta-values expressed in ppm referenced to the CHCl3 residue at 7.25 and 78 ppm, respectively. The FTIR spectra were recorded on a Bruker-Tensor 270 spectrophotometer as KBr disks or as smears between salt plates. The mass spectra were obtained with a GC-Mass Agilent quadrupole mode 5973N instrument, operating at 70 eV.
Cyclic voltammetry measurements were performed on 1 mM solutions of ferrocene derivatives in dry CH3CN/0.1 M LiClO4 using potentiostat/galvanostat Autolab (PGASTAT 30) equipped with a standard three-electrode cell. A 2-mm-diameter GC was used as the working electrode. A silver/silver chloride (Ag/AgCl) electrode was used along with a platinum electrode as the reference and the counter electrodes, respectively. All potentials in this study are reported with respect to the Ag/AgCl reference electrode.
5.3 General procedure for the synthesis of alkylferrocene
A solution of acyl chloride (53.76 mmol) in 25 ml dry dichloromethane was added to a suspension of anhydrous aluminum chloride (7.7 g, 53.76 mmol) in 25 ml dry dichloromethane and the mixture was stirred at 5 °C for 1 h under argon. The solution of aluminum chloride/acyl chloride complex was added dropwise for 30 min to a solution of ferrocene (10 g, 53.76 mmol) in 130 ml dry dichloromethane at 0 °C. The reaction mixture was warmed to room temperature and stirred for 16 h. A solution of NaBH4 (2.29 g, 53.76 mmol) in 25 ml diglyme was added dropwise to the purple reaction mixture at −5 °C to form an orange solution as the result which was stirred for an hour in 0 °C. The mixture was hydrolyzed with water while maintaining its temperature at less than or equal to 10 °C. The mixture was allowed to separate by settling and the organic phase was then withdrawn. The aqueous phase was extracted 3 times with 30 ml of CH2Cl2 and then all the organic phases were combined, washed with 50 ml of brine, CH2Cl2 was removed and the diglyme and the residual ferrocene which was found to be entrained by the diglyme, were distilled at reduced pressure of approximately 20 mm Hg at a column head temperature of 85 °C–95 °C. The alkylferrocene derivatives were distilled, at less than 5 mm Hg. Specific details are given for each compound.
5.3.1 Ethylferrocene (3b)
From 4.16 g acetyl chloride, (10.1 g, 0.047 mol) of light brown liquid was obtained in 89% yield. 1H NMR (400 MHz, CDCl3, ppm): 1.16–1.19 (t, 3H, CH3), 2.32–2.37 (q, 2H, FcCH2), 3.99–4.16 (m, 9H, Fc).
5.3.2 Propylferrocene (3c)
From 4.9 g propionyl chloride, (10.3 g, 0.045 mol) of light brown liquid was obtained in 85% yield. 1H NMR (400 MHz, CDCl3, ppm): 0.93–0.96 (t, 3H, CH3), 1.49–1.58 (m, 2H, CH2CH3), 2.31–2.35 (t, 2H, FcCH2), 4.05–4.15 (m, 9H, Fc).
5.3.3 Butylferrocene (3d)
From 5.64 g butanoyl chloride, (10.8 g, 0.044 mol) of light brown liquid was obtained in 84% yield. 1H NMR (400 MHz, CDCl3, ppm): 0.89–0.92 (t, 3H, CH3), 1.31–1.36 (m, 2H, CH2CH3), 1.42–1.45 (m, 2H, CH2CH2CH3), 2.29–2.36 (t, 2H, CpCH2), 4.02–4.13 (m, 9H, Fc).
5.4 General procedure for the synthesis of (4-chlorobutyl)alkylferrocene
A solution of 4-chlorobutanoyl chloride (7.4 g, 53.76 mmol) in 25 ml dry CH2Cl2 was added dropwise at room temperature to a suspension of anhydrous aluminum chloride (7.7 g, 53.76 mmol) in 25 ml dry CH2Cl2 and stirred at room temperature for 1 h under argon. The homogeneous yellow solution was added dropwise to a solution of ferrocene or alkylferrocene derivatives (53.76 mmol) in 130 ml dry CH2Cl2 at 0 °C. The formed dark purple solution was then allowed to slowly get warm to reach room temperature. After 24 h of stirring at room temperature, a solution of NaBH4 (2.9 g, 53.76 mmol) in 25 ml diglyme was added to the mixture while maintaining the temperature at less than or equal to 0 °C. A dark orange solution was formed as the result and stirred at 0 °C for 1 h. The mixture was hydrolyzed with 60 ml water while keeping its temperature at less than or equal to 5 °C. The mixture was allowed to separate by settling to let the organic phase be withdrawn and the aqueous phase was extracted 3 times with 30 ml of CH2Cl2. All the organic phases were combined and washed with 60 ml of brine. CH2Cl2 was removed and the diglyme was distilled under reduced pressure. The residue was purified by column chromatography on silica gel with n-hexane as the eluent. Specific details are given for each compound.
5.4.1 (4-Chlorobutyl)ferrocene (5a)
From 10 g ferrocene, (13.2 g, 0.047 mol) of orange liquid was obtained in 90% yield. FTIR (KBr, cm−1): 3091 (Cp-H), 2936–2859 (CH), 1640, 1450 (CC), 1305 (CH2Cl)), 1036 (Cp), 820 (CCl), 492 (Cp-Fe); 1H NMR (400 MHz, CDCl3, ppm): 1.54–1.69 (m, 2H, CH2), 1.77–1.81 (m, 2H, CH2), 2.34–2.38 (t, 2H, Cp-CH2), 3.53–3.56 (t, 2H, CH2Cl), 4.05–4.10 (m, 9H, Cp-H); m/z (EI): 276 (100% [M]+), 240 (66.47% [M − HCl]+), 91 (14.82% [CH2CH2CH2CH2Cl]+).
5.4.2 (4-Chlorobutyl)ethylferrocene (5b)
From 11.34 g ethylferrocene, (14.5 g, 0.047 mol) of orange liquid was obtained in 89% yield. FTIR (KBr, cm−1): 3088 (Cp-H), 2860–2931 (CH), 1637, 1450 (CC), 1311 (CH2Cl), 1036 (Cp), 817 (CCl), 490 (Cp-Fe); 1H NMR (400 MHz, CDCl3, ppm): 1.10–1.15 (t, 3H, CH3), 1.65–1.67 (m, 2H, Cp-CH2CH2), 1.77–1.83 (m, 2H, CH2CH2Cl), 2.32–2.37 (m, 4H, Cp-CH2), 3.55–3.56 (t, 2H, CH2Cl), 4.00–4.11 (m, 8H, Cp-H); m/z (EI): 304 (100%[M]+), 91 (26.14% [CH2CH2CH2CH2Cl]+).
5.4.3 (4-Chlorobutyl)propylferrocene (5c)
From 12 g propylferrocene, (15 g, 0.047 mol) of orange liquid was obtained in 88% yield. FTIR (KBr, cm−1): 3091 (Cp-H), 2936–2859 (CH), 1640, 1450 (CC), 1305 (CH2Cl), 1007 (Cp), 817 (CCl), 462 (Cp-Fe); 1H NMR (400 MHz, CDCl3, ppm): 0.93–0.97 (t, 3H, CH3), 1.48–1.57 (m, 2H, CH2CH3), 1.62–1.67 (m, 2H, Cp-CH2CH2), 1.78–1.85 (m, 2H, CH2CH2Cl), 2.25–2.39 (m, 4H, Cp-CH2), 3.53–3.59 (t, 2H, CH2Cl), 3.96–4.12 (m, 8H, Cp-H); m/z (EI): 318 (100%[M]+), 91 (8.54% [CH2CH2CH2CH2Cl]+).
5.4.4 (4-Chlorobutyl)butylferrocene (5d)
From 12.82 g butylferrocene, (15.6 g, 0.046 mol) of orange liquid was obtained in 88% yield. FTIR (KBr, cm−1): 3087 (Cp-H), 2880–2928 (CH), 1685, 1450 (CC), 1307 (CH2Cl), 1036 (Cp), 816 (CCl), 482 (Cp-Fe); 1H NMR (400 MHz, CDCl3, ppm): 0.90–0.98 (t, 3H, CH3), 1.29–1.38 (m, 2H, CH2), 1.43–1.51 (m, 2H, CH2), 1.61–1.68 (m, 2H, Cp-CH2CH2), 1.76–1.83 (m, 2H, CH2CH2Cl), 2.25–2.37 (m, 4H, Cp-CH2), 3.52–3.58 (t, 2H, CH2Cl), 3.97–4.03 (m, 8H, Cp-H); m/z (EI): 332 (100%[M]+) 91 (8.72% [CH2CH2CH2CH2Cl]+).
5.5 General procedure for the synthesis of (3-butenyl)alkylferrocene
A 50 ml round-bottom flask was charged with (4-chlorobutyl)alkylferrocene (7.23 mmol), sodium tert-butoxide (2.0 g, 21.0 mmol) and 30 ml DMSO as the solvent under argon. Mixture was stirred at 60 °C and reaction progress was monitored by thin layer chromatography (TLC) until the complete disappearance of (4-chlorobutyl)alkylferrocene. The mixture was allowed to cool to room temperature to get ready for being extracted three times with 30 ml of n-hexane. All the n-hexane phases were combined and washed 3 times with 10 ml of water to remove sodium tert-butoxide and DMSO. The organic phase was dried over Na2SO4 and the organic solvent was removed. The residue was purified by column chromatography with n-hexane as the eluent to give the corresponding products (Rf = 0.6). Specific details are given for each compound.
5.5.1 (3-Butenyl)ferrocene (6a)
From 2 g of (4-chlorobutyl)ferrocene, 1.5 g (6.25 mmol) of brown oil (88% yield) was obtained. FTIR (KBr, cm−1): 3087 (CH), 2926, 2860 (CH), 1637, 1451 (CC), 1101, 1009, (Cp); 1H NMR (400 MHz, CDCl3): δ 2.27–2.32 (m, 2H, CH2), 2.42–2.46 (t, 2H, CH2), 4.06–4.12 (m, 9H, Cp-H), 4.98–5.08 (dd, 2H, CHCH2), 5.85–5.92 (ddt, 1H, CHCH2); m/z (EI): 240 (80.78% [M]+), 199(100% [Fc-CH2]+), 121 (43.5% [Cp-Fe]+) [32].
5.5.2 (3-Butenyl)ethylferrocene (6b)
From 2.2 g of (4-chlorobutyl)ethylferrocene, 1.5 g (5.5 mmol) of brown oil (88% yield) was obtained. FTIR (KBr, cm−1): 3087 (CH), 2926, 2860 (CH), 1637, 1451 (CC), 1101, 1000, (Cp); 1H NMR (400 MHz, CDCl3): δ 1.14–1.17 (t, 3H, CH3), 2.25–2.42 (m, 6H, CH2), 3.97–4.10 (m, 8H, Cp-H), 4.96–4.98 (dd, 2H, CHCH2), 5.86–5.87 (ddt, 1H, CHCH2); 13C NMR (100 MHz, CDCl3, ppm): 13.87 (CH3), 21.13 (CH2), 28.08 (Fc-CH2), 34.43 (CH2), 66.70, 67.45, 67.98 (Cp), 87.02 (Cipso), 113.39 (CHCH2), 137.62 (CHCH2); m/z (EI): 268 (91.5% [M]+), 227 (100% [CH3CH2Fc-CH2]+).
5.5.3 (3-Butenyl)propylferrocene (6c)
From 2.3 g of (4-chlorobutyl)propylferrocene, 1.5 g (5.3 mmol) of brown oil (75% yield) was obtained. FTIR (KBr, cm−1): 3083 (CH), 2926, 2885 (CH), 1637, 1453 (CC), 1102, 1026, (Cp); 1H NMR (400 MHz, CDCl3): δ 0.91–0.95 (t, 3H, CH3), 1.48–1.52 (t, 2H, CH2), 2.27–2.29, 2.36–2.42 (m, 6H, CH2), 3.99–4.11 (m, 8H, Cp-H), 4.96–5.06 (dd, 2H, CHCH2), 5.86–5.90 (ddt, 1H, CHCH2); 13C NMR (100 MHz, CDCl3, ppm): 13.25 (CH3), 23.46 (CH2), 28.04, 30.75 (CH2), 34.42 (CH2), 66.71, 67.46, 67.99 (Cp-H), 87.47 (Cipso), 113.45 (CHCH2), 137.70 (CHCH2); m/z (EI): 282 (100% [M]+), 241(42.8% [CH3(CH2)2Fc-CH2]+).
5.5.4 (3-Butenyl)butylferrocene (6d)
From 2.4 g of (4-chlorobutyl)butylferrocene, 1.5 g (5.06 mmol) of brown oil (70% yield) was obtained. FTIR (KBr, cm−1): 3083 (CH), 2926, 2858 (CH), 1637, 1453 (CC), 1103, 1026, (Cp); 1H NMR (400 MHz, CDCl3): δ 0.92 (t, 3H, CH3), 1.34–1.47 (m, 4H, CH2), 2.28–2.41 (m, 6H, CH2), 3.98–4.03 (m, 8H, Cp-H), 4.98–5.06 (dd, 2H, CHCH2), 5.87 (ddt, 1H, CHCH2); 13C NMR (100 MHz, CDCl3, ppm): 13.02 (CH3), 21.65 (CH2), 27.93, 28.07 (Fc-CH2), 32.45, 34.41 (CH2), 65.79, 76.56, 68.01(Cp-H), 87.48 (Cipso), 113.45 (CHCH2), 137.71 (CHCH2); m/z (EI): 296 (100% [M]+), 255(78% [CH3(CH2)3Fc-CH2]+), 121 (15% [Cp-Fe]+).
5.6 Preparation of [4-(alkylferrocenyl)butyl]dimethylsilane
5.6.1 (4-Ferrocenylbutyl)dimethylsilane (7a)
7a was prepared according to the procedure described by M. Immelman, J.C. Swarts, G.J. Lamprecht, and S.E. Greyling [30].
5.6.2 [4-(Ethylferrocenyl)butyl]dimethylsilane (7b)
7b was prepared according to a similar procedure to that of 7a as dark brown oil in 91% yield: FTIR (KBr, cm−1): 3089 (Cp-H), 2960 (CH), 2110 (SiH), 1635, 1454 (Cp), 1250 (CSi), 1031 (Cp), 491 (Cp-Fe); 1H NMR (400 MHz, CDCl3, ppm): 0.09–0.14 (d, 6H, Si(CH3)2), 0.61–0.65 (t, 2H, CH2Si(CH3)2), 0.91–0.96 (m, 2H, CH2CH2Si(CH3)2), 1.16–1.21 (t, 3H, CH3), 1.43–1.56 (m, 2H, Cp-CH2CH2), 2.31–2.37 (m, 4H, Cp-CH2), 3.97–4.05 (m, 9H, Cp, SiH); 13C NMR (100 MHz, CDCl3, ppm): −3.59 (Si(CH3)2), 13.26 (CH2CH3), 25.36 (CH2Si(CH3)2), 27.88, 28.49 (CH2), 31.56, 34.65 (CpCH2), 66.58, 67.13, 67.31, 67.38 (Cp), 86.14, 88.28 (CH2C5H3); Anal. Calc. for: C18H28FeSi (328.347): C, 65.84; H, 8.59; Fe, 17.01. Found: C, 63.99; H, 8.33%; Fe, 16.94%.
5.7 General procedure for the synthesis of bis[4-(alkylferrocenyl)butyl]dimethylsilane derivatives (8a–g)
A 25 ml round-bottom flask with a magnetic stirrer was charged with (3-butenyl) alkylferrocene (0.83 mmol), [(4-dimethylsilyl)butyl]ferrocene or [(4-dimethylsilyl)butyl]ethylferrocene (1.2 mmol) and 15 ml dry toluene solvent. Following this, 10 μl of Karstedt catalyst ([Pt]/[SiH] = 3.1 × 10−6) was added to the mixture. FTIR spectroscopy was utilized to follow the progress of the reaction, by monitoring the loss of the SiH absorption. The reaction mixture was stirred at room temperature until the complete disappearance of SiH bonds in FTIR spectra. The solvent was evaporated under reduced pressure and the residue was purified by column chromatography with (20 ml) n-hexane as the eluent to give the corresponding products. Specific details are given for each compound.
5.7.1 Bis(4-ferrocenylbutyl)dimethylsilane (8a)
From 0.2 g of 3-butenylferrocene and 0.37 g of (4-ferrocenylbutyl)dimethylsilane, 0.45 g (0.83 mmol) of orange oil (91% yield) was obtained.
FTIR (KBr, cm−1): 3090 (CH), 2921, 2795 (CH), 1633, 1410 (CC), 1252, 814 (SiC), 1101, 1003 (Cp); 1H NMR (400 MHz, CDCl3): δ −0.0175 (s, 6H, Si-(CH3)2), 0.50–0.54 (t, 4H, CH2Si), 1.34–1.36 (m, 4H, CH2), 1.51–1.55 (m, 4H, CH2), 2.30–2.34 (t, 4H, CH2), 4.06–4.17 (m, 18H, Cp-H); 13C NMR (100 MHz, CDCl3, ppm): −4.07 (Si-(CH3)2), 13.86, 22.91, 28.18, 33.99 (CH2), 65.99, 66.88, 67.39 (Cp-H), 88.56 (Cipso).
5.7.2 [4-(Ethylferrocenyl)butyl](4-ferrocenylbutyl)dimethylsilane (8b)
From 0.22 g of (3-butenyl)ethylferrocene and 0.37 g of (4-ferrocenylbutyl)dimethylsilane, 0.43 g (0.75 mmol) of orange oil (91% yield) was obtained. FTIR (KBr, cm−1): 3087 (CH), 2922, 2850 (CH), 1710, 1452(CC), 1252, 819 (SiC), 1101, 1003 (Cp); 1H NMR (400 MHz, CDCl3): δ −0.031 (s, 6H, Si-(CH3)2), 0.49–0.53 (t, 4H, CH2Si), 1.14–1.16 (t, 3H, CH3), 1.31–1.35 (m, 4H, CH2), 1.49–1.54 (m, 4H, CH2), 2.30–2.33 (m, 6H, CH2-Fc), 3.98–4.09 (m, 17H, Cp-H). 13C NMR (100 MHz, CDCl3, ppm): −4.30 (Si-(CH3)2), 13.83 (CH3), 21.17 (CH2), 22.95, 22.99, 28.69, 34.01 (CH2), 65.98, 66.42, 67.03, 67.46 (Cp-H), 88.56 (Cipso).
5.7.3 (4-Ferrocenylbutyl)[4-(propylferrocenyl)butyl]dimethylsilane (8c)
From 0.23 g of (3-butenyl)propylferrocene and 0.37 g of (4-ferrocenylbutyl)dimethylsilane, 0.42 g (0.72 mmol) of yellow oil (87% yield) was obtained. FTIR (KBr, cm−1): 3086 (CH), 2923, 2850 (CH), 1642, 1456(CC), 1260, 814 (SiC), 1101, 1003 (Cp); 1H NMR (400 MHz, CDCl3): δ −0.02 (s,6H, Si-(CH3)2), 0.54–0.56 (t,4H, CH2Si), 0.96–1.00 (t, 3H, CH3), 1.31–1.37 (m, 4H, CH2), 1.55–1.57 (m, 6H, CH2), 2.30–2.36 (t, 6H, CH2-Fc), 3.92–4.14 (m, 17H, Cp-H). 13C NMR (100 MHz, CDCl3, ppm): −4.30 (Si-(CH3)2), 14.16 (CH3), 22.93, 23.45, 28.06, 28.21 (CH2), 33.99, 34.20 (CH2-Fc), 65.93, 67.01, 67.63, 67.97 (Cp-H), 88.50 (Cipso).
5.7.4 [4-(Butylferrocenyl)butyl](4-ferrocenylbutyl)dimethylsilane (8d)
From 0.24 g of (3-butenyl)butylferrocene and 0.37 g of (4-ferrocenylbutyl)dimethylsilane, 0.40 g (0.67 mmol) of yellow oil (81% yield) was obtained. FTIR (KBr, cm−1): 3088 (CH), 2923, 2858 (CH), 1708, 1454(CC), 1255, 818 (SiC), 1101, 1003 (Cp); 1H NMR (400 MHz, CDCl3): δ −0.029 (s, 6H, Si-(CH3)2), 0.51–0.53 (t, 4H, CH2Si), 0.90–0.93 (t, 3H, CH3), 1.31–1.35 (m, 6H, CH2), 1.49–1.53 (m, 6H, CH2), 2.30–2.31 (m, 6H, CH2-Fc), 3.97–4.10 (m, 17H, Cp-H); 13C NMR (100 MHz, CDCl3, ppm):−4.30 (Si-(CH3)2), 14.14 (CH3), 22.91, 28.19, 32.14, 34.16 (CH2), 65.94, 66.75, 67.41, 67.70 (Cp-H), 88.49 (Cipso).
5.7.5 Bis[4-(ethylferrocenyl)butyl]dimethylsilane (8e)
From 0.22 g of (3-butenyl)ethylferrocene and 0.4 g of [4-(ethylferrocenyl)butyl]dimethylsilane, 0.41 g (0.68 mmol) of yellow oil (83% yield) was obtained. FTIR (KBr, cm−1): 3087 (CH), 2922, 2850 (CH), 1710, 1452(CC), 1252, 819 (SiC), 1103, 1056 (Cp); 1H NMR (400 MHz, CDCl3): δ −0.002 (s,6H, Si-(CH3)2), 0.52–0.55 (t, 4H, CH2Si), 1.16–1.21 (t, 6H, CH3), 1.35–1.37 (m, 4H, CH2), 1.52–1.55 (m, 4H, CH2), 2.31–2.37 (m, 8H, CH2-Fc), 3.97–4.12 (m, 16H, Cp-H); 13C NMR (100 MHz, CDCl3, ppm): −4.31(Si-(CH3)2), 14.11 (CH3), 21.08 (CH2Si), 22.90, 22.97, 28.06, 34.23 (CH2), 66.52, 66.93, 67.47, 67.89 (Cp-H), 88.27 (Cipso).
5.7.6 [4-(Ethylferrocenyl)butyl][4-(propylferrocenyl)butyl]dimethylsilane (8f)
From 0.23 g of (3-butenyl)propylferrocene and 0.4 g of [4-(ethylferrocenyl)butyl]dimethylsilane, 0.43 g (0.7 mmol) of yellow oil (86% yield) was obtained. FTIR (KBr, cm−1): 3086 (CH), 2923, 2850 (CH), 1642, 1456 (CC), 1260, 814 (SiC), 1097, 1017 (Cp); 1H NMR (400 MHz, CDCl3): δ −0.004 (s, 6H, Si-(CH3)2), 0.53 (t, 4H, CH2Si), 0.96–1.181 (t, 6H, CH3), 1.183–1.196 (m, 4H, CH2), 1.52–1.54 (m, 6H, CH2), 2.30–2.35 (m, 8H, CH2-Fc), 3.98–4.06 (m, 16H, Cp-H); 13C NMR (100 MHz, CDCl3, ppm): −4.30 (Si-(CH3)2), 14.14 (CH3), 22.96 (CH2Si), 28.07, 28.22, 30.62, 33.97, 34.21 (CH2), 66.96, 67.51, 67.88 (Cp-H), 88.11(Cipso).
5.7.7 [4-(Butylferrocenyl)butyl][4-(ethylferrocenyl)butyl]dimethylsilane (8g)
From 0.24 g of (3-butenyl)butylferrocene and 0.4 g of [4-(ethylferrocenyl)butyl]dimethylsilane, 0.41 g (0.65 mmol) of yellow oil (80% yield) was obtained. FTIR (KBr, cm−1): 3086 (CH), 2923, 2860 (CH), 1695, 1442(CC), 1260, 814 (SiC), 1097, 1017 (Cp); 1H NMR (400 MHz, CDCl3): δ −0.027 (s, 6H, Si-(CH3)2), 0.53 (t, 4H, CH2Si), 0.92–0.94, 1.17 (t, 6H, CH3), 1.33–1.37 (m, 6H, CH2), 1.50–1.53 (m, 6H, CH2), 2.29–2.35 (m, 8H, CH2-Fc), 3.97–4.10 (m, 16H, Cp-H); 13C NMR (100 MHz, CDCl3, ppm): −4.30 (Si-(CH3)2), 14.18 (CH3), 22.66, 22.91, 22.98, 28.08, 33.89, 34.21 (CH2), 66.60, 67.41, 67.97 (Cp-H), 88.39 (Cipso).
5.8 Synthesis of symmetrical bis[4-(alkylferrocenyl)butyl]dimethylsilane derivatives (8a and 8e) by Grignard reaction
The Grignard reagent was prepared in situ by adding of 20 ml of a solution of 4-chlorobutylalkylferrocene derivatives 5a or 5b (18 mmol) in dry THF (80 ml) to 10 ml dry THF containing magnesium turnings (0.43 g, 18 mmol) and a little iodine crystal. The mixture was stirred at room temperature until the reaction commenced (15 min), after which the remaining 60 ml of the chloro-containing compound in THF solution was added dropwise as the reaction went on. The reaction mixture was refluxed under nitrogen until all the magnesium dissolved. The cooled THF-solution containing the Grignard reagent was subsequently added under nitrogen in 30 min, to an excess of dichlorodimethylsilane (1.5 g, 11.6 mmol) and was refluxed for 16 h. After filtration of the precipitated MgCl2, the solvent was evaporated and the residue was purified by column chromatography with n-hexane as the eluent to give the corresponding products. Specific details are given for each compound.
5.8.1 Bis(4-ferrocenylbutyl)dimethylsilane (8a) from Grignard reaction
From 5 g of 4-chlorobutylferrocene (5a), 4.48 g (8.3 mmol) of orange oil (92% yield) was obtained with specific details like in hydrosilylation product 8a.
5.8.2 Bis[4-(ethylferrocenyl)butyl]dimethylsilane (8e) from Grignard reaction
From 5.48 g of 4-chlorobutylethylferrocene (5a), 3.67 g (7.6 mmol) of orange oil (84% yield) was obtained with specific details like in hydrosilylation product 8e.
Acknowledgements
The authors would like to thank the financial support of the Center of Excellence in Clean Chemistry and New Materials of Faculty of Chemistry, University of Tabriz, Tabriz, Iran.