

1 Introduction
Biaryls are a key structural feature of numerous natural products, biologically active molecules, tools for asymmetric synthesis, pharmaceuticals, agrochemicals, and other materials (Fig. 1). Various approaches have been developed toward efficient methods for their atroposelective synthesis [1]. Three important issues have to be addressed for their synthesis, namely, the regio- and stereoselective control in biaryl synthesis and the minimization of metal amounts in these processes.
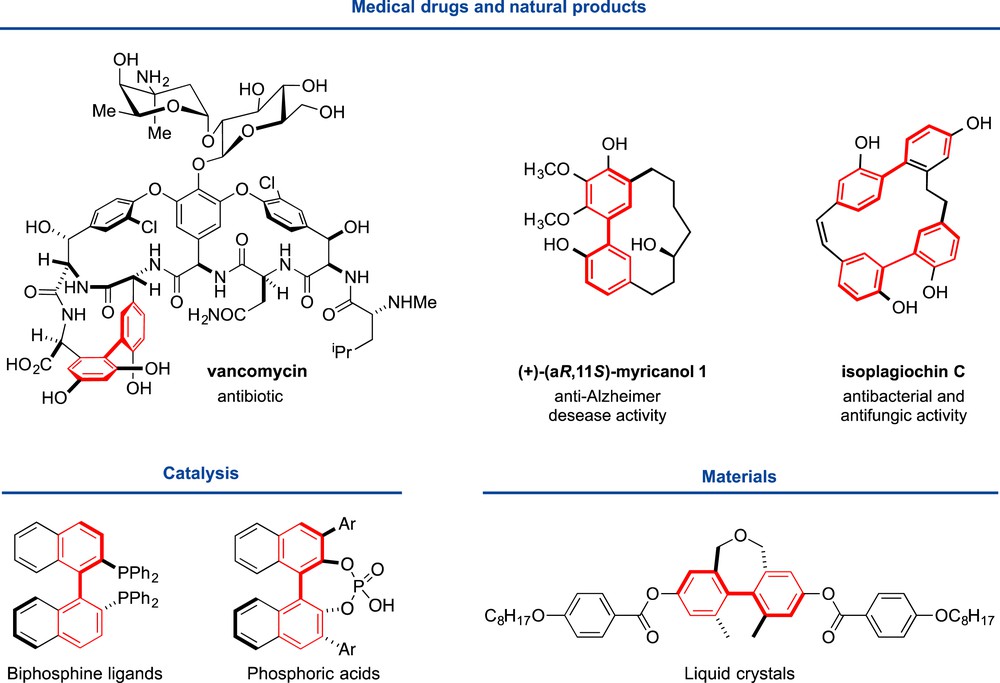
Biaryls as important structural motifs.
In recent years, classical methods for creating aryl–aryl bonds [2] have been complemented by direct arylation with the carbonhydrogen bond used as a functional group (Fig. 2) [3]. The major drawback in these approaches is the use of heavy metals, which may cause contamination of the products, requiring purification for biological applications. Because of potentially toxic contamination of pharmaceutical products, effective removal of the metal (Pt, Pd, Ir, Rh, Ru, or Os) in active pharmaceutical ingredients causes acute problems for pharmaceutical companies [4].

Access routes toward biaryls.
In the present work, we report on our contributions in this field. The transition metal-free ARYNE coupling will be presented toward first racemic biaryl scaffolds and then our developments for the synthesis of enantiopure or -enriched biaryls.
2 Results and discussion
2.1 Aryne route to access biaryls
The use of arynes to access biaryls or polyaryls has been nicely reviewed recently [5]. The first example of such a process was described by Wittig et al. [6] in 1940 using phenyllithium and fluorobenzene. Later, Gilman and colleagues [7] in 1950s reported on the reaction of ortho-dihalobenzenes with butyllithium leading to a biphenyl backbone, presumably via a 1,2-didehydroarene, that is, an ortho-aryne (Scheme 1). This halogen/lithium interconversion-based approach remained dormant for several decades except for a procedure improvement [8].

Gilman's observation on the reaction of ortho-dihalobenzenes with butyllithium. Upper line with stoichiometric amounts and lower line with half a molar equivalent.
We started reinvestigating this reaction in 2001 and then extended its scope to access various biaryls, and we clarified the mechanism of the reaction. The reaction of aryllithium compounds with ortho-dihaloarenes, proceeding via the in situ formation of arynes allows the preparation of ortho-halobiaryls. This “ARYNE coupling” has become a robust method for aryl–aryl coupling and combines several advantages, as the use of cheap and/or easily accessible halogen aromatic compounds, the access to biaryls bearing two distinct aromatic units and to polyhalogenated biaryls that can be functionalized further, the use of lithium reagents (i.e., in the absence of transition metals), and multigram reaction scales (Fig. 3) [9].
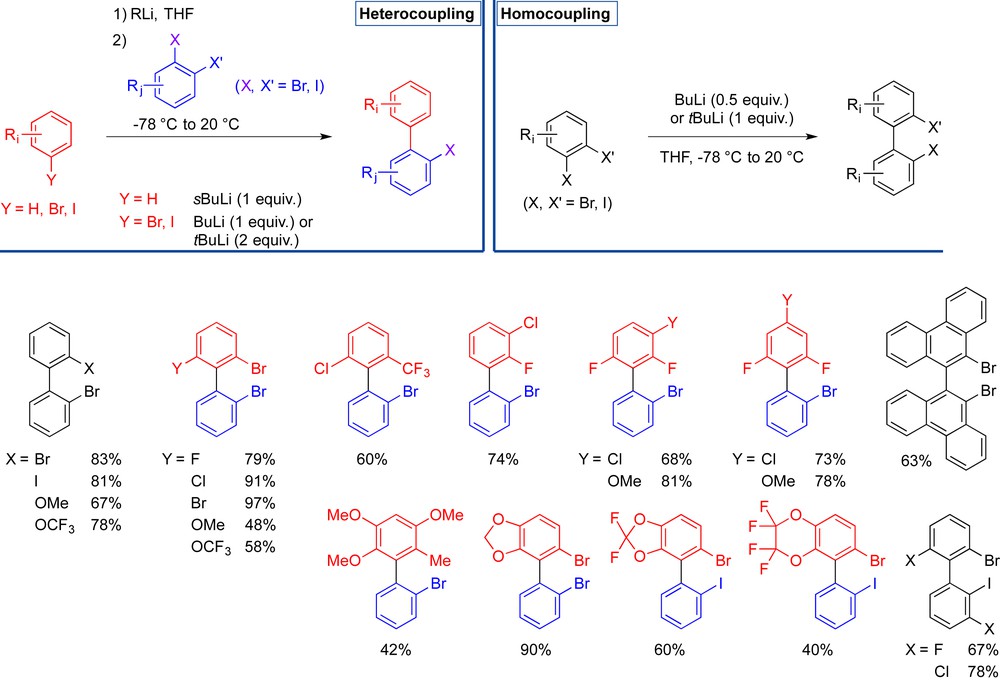
ARYNE coupling toward ortho, ortho′-, di-, tri- and tetra-substituted biaryls.
The mechanism is based on a subtle interplay of several organometallic species and their relative basicities (Fig. 4). The chain reaction proceeds as follows: (1) A thermodynamically stable organolithium intermediate is formed, of which (2) a small amount performs a halogen/metal exchange on the coupling partner via an ate complex, generating a thermodynamically unstable ortho-halophenyllithium intermediate (initiation). The latter (3) eliminates spontaneously lithium halide affording an aryne; then, (4) the transient aryne species undergoes nucleophilic addition of the organolithium precursor; and (5) the resulting 2-biaryllithium intermediate is finally stabilized by in situ transfer of bromine or iodine from the starting material, affording again some ortho-halophenyllithium to pursue the chain reaction (propagation steps). The termination of the chain reaction proceeds by hydrolysis of aryllithium species (by abstraction of a proton of the solvent or during aqueous work up) and by dimerization or trimerization of residual aryne traces (the corresponding side products being sometimes detected by gas chromatography mass spectrometry (GC–MS) analysis).

Mechanism of the ARYNE coupling.
The next logical step was then to apply this ARYNE coupling to the preparation of axially chiral biaryls. The synthesis of axially stereoenriched biaryls remains a challenging and extensively studied research field because of the above-mentioned importance of the biaryl motif. The most important approaches are the resolution of preconstructed biaryls and the atroposelective coupling of two aromatic partners [1a–f,10a–c].
We first focused on the resolution strategy by taking advantage of a specificity of the ARYNE coupling, namely, the regeneration of a carbon-exchangeable halogen bond at position 2 of the biaryl product, which can be further functionalized.
2.2 Regioselective halogen/metal exchange
Systematic investigations carried out in our laboratory have shown that differences exist in the stabilities of aryllithium intermediates generated by butyllithium-promoted halogen/metal interconversion on bromobenzene and heterosubstituted close congeners. For example, in an intramolecular competition experiment, we could show that 1,3-dibromo-5-[(3-bromophenylmethoxy)methyl]benzene underwent exclusively a halogen/metal permutation with butyllithium at the multiple substituted ring (Scheme 2).
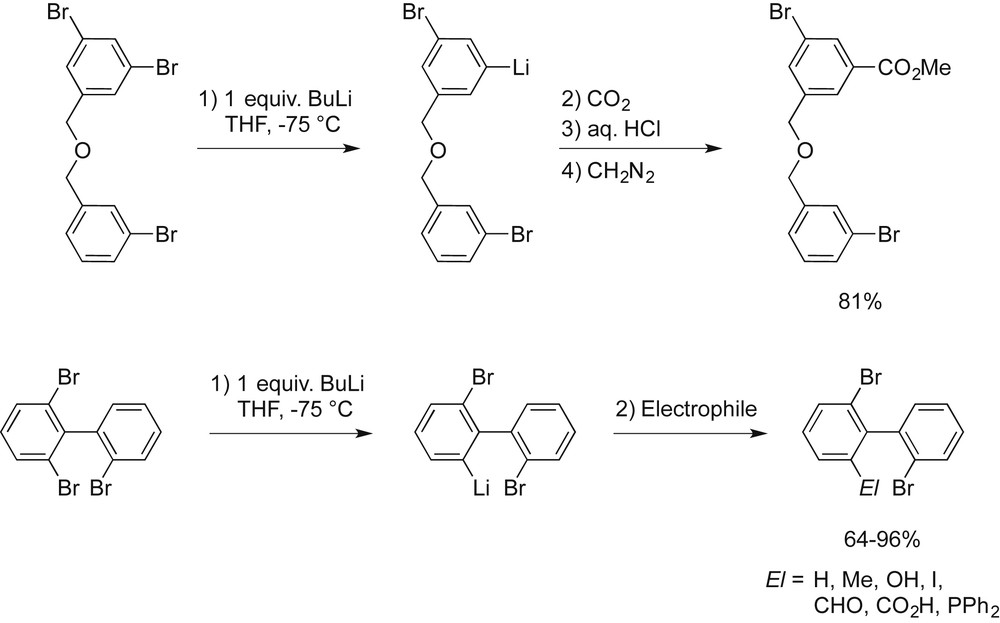
Intramolecular competition experiments revealing a regioselective bromine/lithium interconversion.
Similarly, 2,2′,6-tribromobiphenyl obtained on the multigram scale via the ARYNE coupling in almost quantitative yield [9c,11] undergoes successive and completely regioselective bromine/lithium interconversion starting on the higher substituted phenyl ring [12]. Such an effective discrimination between two bromine atoms as a function of their chemical environment has so far been observed only sporadically in such kind of processes [13].
Thus, on the basis of ARYNE coupling, giving rise to a large number of diversely substituted coupling products, and the regioselective bromine/lithium interconversion and trapping with various electrophiles, we were able to access a multitude of functionalized biaryls in a modular way [9d,14].
2.3 Post-ARYNE coupling desymmetrization or deracemization of achiral biaryls
The regioselectivity issue being addressed, the next step was then to control the axial chirality. Therefore, we decided to introduce after the first bromine/lithium exchange a traceless chiral auxiliary, namely the para-toluenesulfinyl group. In fact, the sulfinyl group presents numerous synthetic advantages [15]: (1) its high optical stability; (2) the existence of a large number of sulfinylating agents in both enantiomeric forms, (3) its well-known ortho directing ability and the efficiency as a carrier of the chiral information, (4) its electronic properties, which enhance the contrast between the physical properties of the two stereoisomers, mainly because of dipole orientation [15a], (5) it is very useful as traceless resolving agents, because it may undergo sulfoxide/lithium exchange affording new organometallic intermediates, which can subsequently be trapped by various electrophiles [12a,16a,b].
When we submitted 2,2′,6-tribromobiphenyl or other analogues to the regioselective bromine/lithium interconversion [12b,c], followed by trapping with enantiomerically pure (1R,2S,5R)-(−)-menthyl-(S)-p-toluenesulfinate [17], the atropodiastereomeric biaryl sulfoxides were obtained in excellent yields. Simple crystallization allowed for the separation of both atropodiastereoisomers (S,aR) and (S,aS) in 54% and 70% yield based on the theoretically possible yield of each diastereoisomer (Scheme 3).

Desymmetrization of achiral or deracemization of racemic ortho-dibromobiphenyls by enantiomerically pure p-tolyl sulfoxide as chiral auxiliary.
The most challenging question was whether a chemoselective functionalization of the atropodiastereomerically pure biaryls is possible without any racemization. Both substituents, the sulfoxide group and the bromine atoms, may undergo exchange reactions. We observed with standard organolithium reagents (butyllithium or tert-butyllithium) no chemoselectivity between both types of substituents. In contrast, when using P. Knochel's iPrMgCl·LiCl reagent, a clean sulfoxide/magnesium interconversion occurred at −50 °C. Unfortunately, when the intermediate biaryl Grignard reagent was treated with an electrophile, the temperature had to be raised to 0 °C, which caused partial racemization of the biaryl axis. Thus, we had to find an exchange reagent, which (1) allows the discrimination between the sulfoxide group and the bromine atom(s) and (2) which leads to an intermediate that is reactive enough for trapping at lower temperature. Phenyllithium appeared as the reagent of choice to fulfill all these requirements.
When atropodiastereomeric biaryl sulfoxides were treated with PhLi at −78 °C, a clean sulfoxide/Li interconversion occurred selectively within 10 min. Subsequent trapping with various electrophiles afforded enantioenriched biaryls with excellent er's (≥96:4, Scheme 4). Furthermore, because two atropodiastereomers were initially generated upon sulfinylation, this method allowed to access both enantiomers of the same biphenyl with identical yield and enantiopurity after the sulfoxide/lithium exchange-electrophilic trapping sequence (see the enantiomers of 2,2′-dibromo-6-methylbiphenyl in Scheme 4).
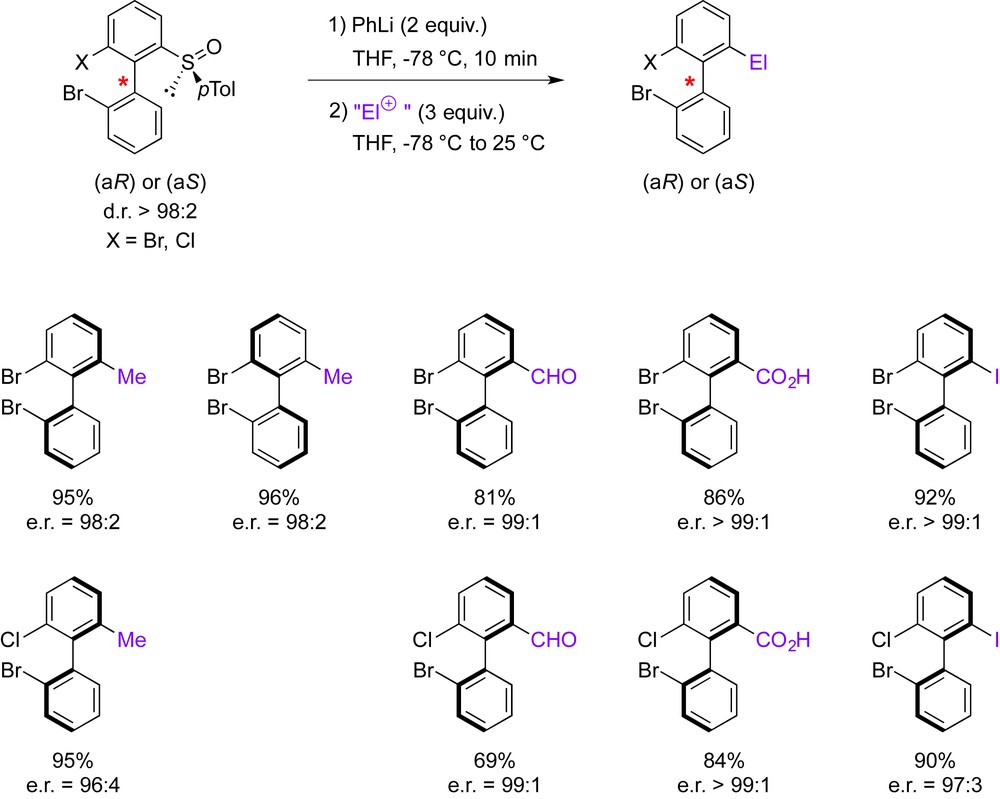
Chemoselective functionalization of atropoisomerically pure biaryl sulfoxides.
To show the scope and the modularity of this approach, we decided to perform a complete functionalization of the biaryl via subsequent sulfoxide and bromine/lithium interconversions and to introduce three types of different substituents. We succeeded in functionalizing 2,2′,6-tribromobiphenyl and to introduce a methoxy-, a methyl group, and a carboxylic function after prior desymmetrization according to the sulfoxide protocol (Scheme 5). A slight erosion of enantiopurity was noticed in the last bromine/lithium exchange and was expected because of the small size of all ortho-substituents on the corresponding biphenyllithium intermediate [18]. Yet the strategy was validated and enantiopure biphenyls could be obtained from 2,2′,6-tribromobiphenyl after sequential element/lithium exchanges provided that the new substituents introduced are bulky enough.
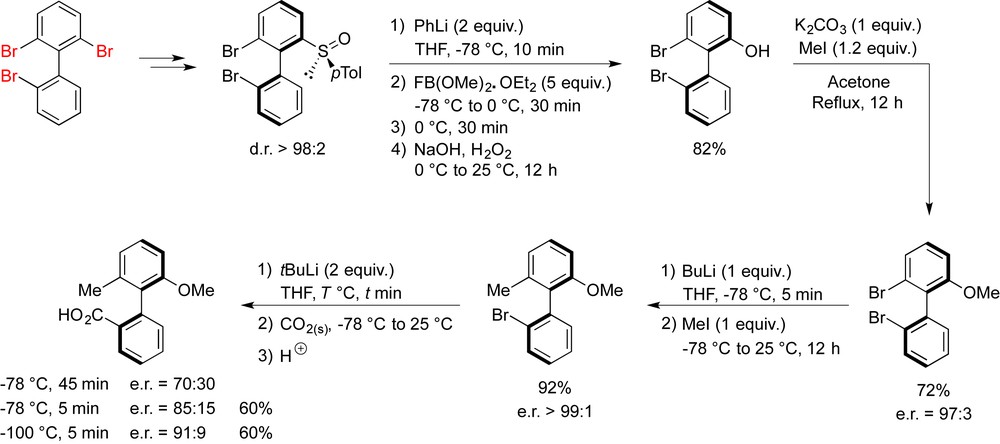
Modular atropoenantiopure biaryl construction starting initially from achiral 2,2′,6-tribromobiphenyl.
2.4 Atropodiastereoselective ARYNE coupling
We next wondered if an atropodiastereoselective ARYNE coupling, where a coupling partner bears a chiral auxiliary before the coupling, might be possible allowing to yield atropodiastereomers of the desired biaryl [9i,j,l]. The choice of the chiral auxiliary and its location onto the coupling partners revealed to be particularly critical. First, the auxiliary should be able to bind to lithium to produce more rigid transition states and, consequently, lead to higher stereoselectivity. Therefore, an oxygen- and/or nitrogen-bearing auxiliary was needed. Second, to avoid regioselectivity issues during the addition of the aryllithium nucleophile onto the aryne, the auxiliary should be located on the former, not on the latter.
2.4.1 Aryl tert-butyl sulfoxides
Our first attempt toward the atropodiastereoselective ARYNE coupling used aryl tert-butyl sulfoxides [9i]. After a preliminary study with model substrates prepared from 3-chloro or 3-methoxyphenyllithium with (S)-di-tert-butyl thiosulfinate, we soon realized that the usual ARYNE coupling conditions (1 equiv of BuLi to generate the aryllithium nucleophile, followed by addition of 1 equiv of a 1,2-dibromobenzene as aryne precursor) were ineffective to produce the expected biaryls. We assumed a low reactivity of the 2-lithioaryl sulfoxide and/or of the 2-lithio-2′-(tert-butylsulfinyl)-1,1′-biphenyl in the halogen/lithium exchange reaction with the brominated aryne precursor, thus blocking the initiation and/or the propagation of the chain reaction, respectively (Fig. 5).
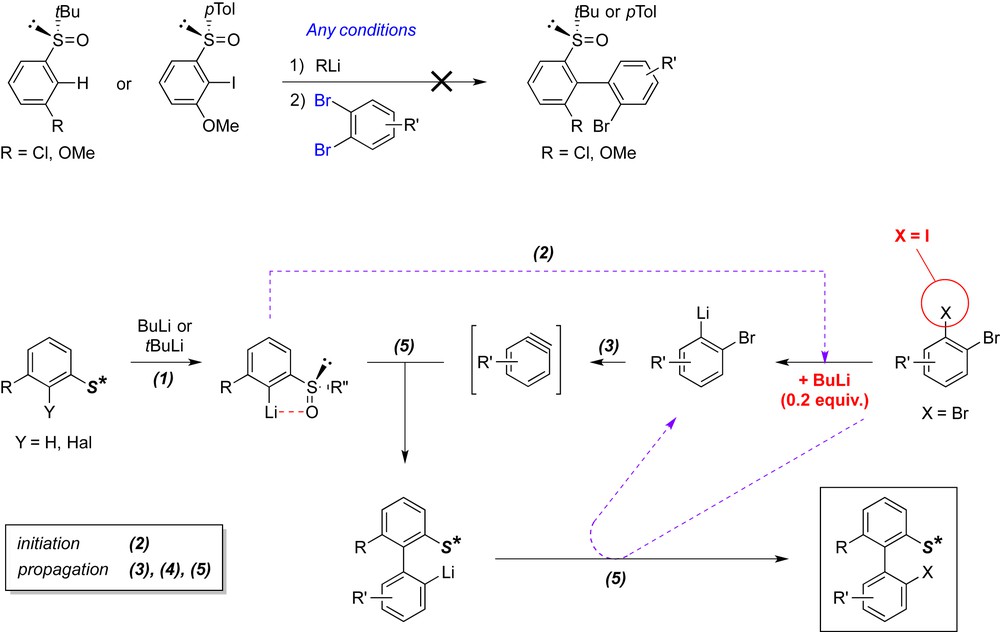
Attempts of atropodiastereoselective ARYNE coupling based on tert-butyl sulfoxides.
This problem could be solved by replacing the dibrominated aryne precursors with the corresponding 1-bromo-2-iodobenzenes and adding a small amount of additional BuLi after introduction of the aryne precursor so as to trigger the generation of the aryne. However, it appeared difficult to achieve simultaneously high yield and reasonable diastereoselectivity (see Scheme 6).
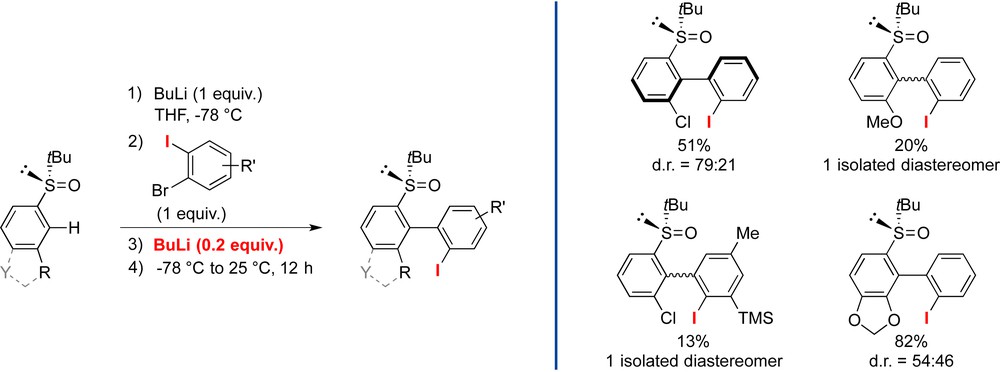
Improved coupling conditions for the atropodiastereoselective ARYNE coupling with tert-butyl sulfoxides.
2.4.2 Aryl para-tolyl sulfoxides
We then considered aryl p-tolyl sulfoxides as appealing alternatives in the atropodiastereoselective ARYNE coupling. Unlike the corresponding tert-butyl sulfoxides, aryl p-tolyl sulfoxides can be easily converted into diversely functionalized arenes by sulfoxide/metal exchange using lithium or magnesium bases (for leading reviews, see Refs. [12a,16a,b,19])—a valuable asset for the derivatization of biaryls, as others and we already demonstrated [9g,20]. Unfortunately, application of the procedure developed for tert-butyl sulfoxides (vide supra) [9i] proved inefficient. Consequently, we forced the conditions by following the introduction of 1-bromo-2-iodobenzene into the solution of aryllithium by the addition of a second equivalent of BuLi. We expected a higher amount of aryne electrophile to be produced to increase the trapping of the ortho-lithioaryl sulfoxide. The reaction mixture was allowed to stir for 12 h before addition of iodomethane to trap the expected biaryllithium intermediate. However, the desired biaryl was not observed but instead an unexpected ortho-terphenyl, apart from the methylated aryl sulfoxide (Scheme 7). A possible explanation for its formation is depicted in Fig. 6. Internal attack of the biaryllithium onto the sulfinyl group would lead, via an oxysulfurane intermediate, to a ligand coupling reaction [19], possibly by 1,2-migration of the tolyl group from sulfur to the arylic carbon in an SNAr fashion, providing a sulfenate anion, which would finally be trapped by iodomethane.

Diverging course of the atropodiastereoselective ARYNE coupling with the p-tolylsulfinyl group as chiral auxiliary.
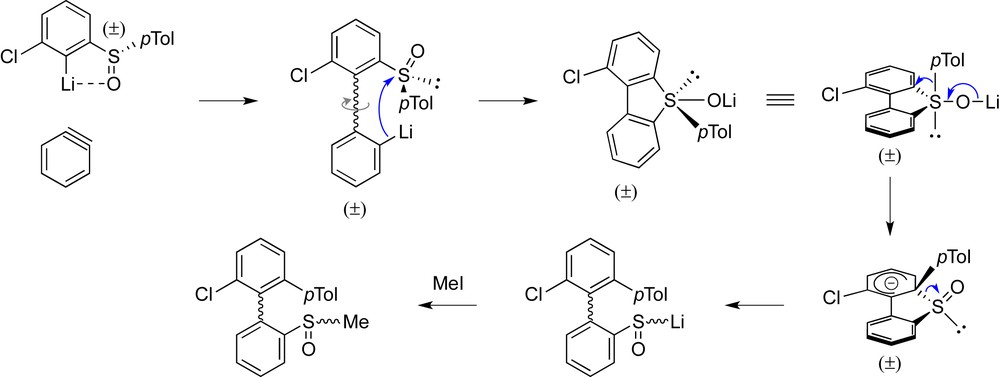
Tentative explanation for the formation of tolyl migration product.
2.4.3 Aryl diethers
Besides the use of sulfur-based chiral auxiliaries, we also studied oxygen-based ones like chiral diethers derived from hydrobenzoin or tartaric acid, inspired by the work of Lipshutz and colleagues [21] on atroposelective intramolecular oxidative aryl–aryl couplings.
When we opposed a tartrate-derived alkoxyaryl iodide to ortho-bromoiodobenzenes (Scheme 8), we were pleased to obtain the desired coupling products in good yields. The compounds were obtained with modest diastereomeric ratios of 60:40 and 62:38. The moderate diastereomeric ratios in comparison with tert-butyl sulfoxide could be caused by the dynamic structure of the aryllithium and biaryllithium intermediates, which could exist as different species in equilibrium, where the tartrate-derived polyether auxiliary could coordinate lithium through different oxygen atoms.
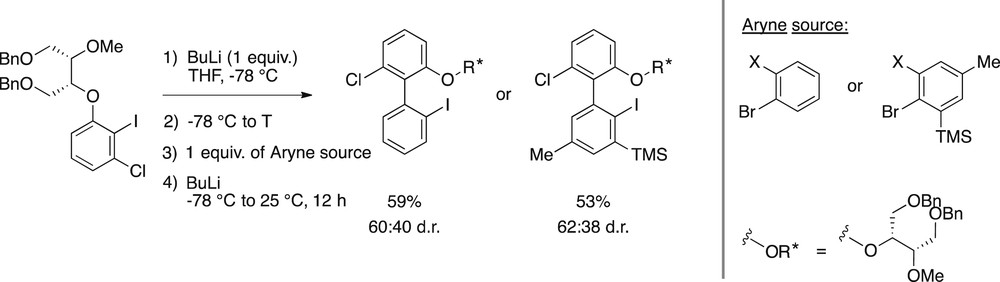
Atropodiastereoselective coupling of chiral diether-bearing substrates.
2.4.4 Aryloxazolines
After these studies on sulfur- and oxygen-based chiral auxiliaries for the atropodiastereoselective ARYNE coupling, we studied chiral oxazolines. The oxazoline group is a well-known chiral inductor and a well-known directing group in polar organometallic chemistry [22]. Moreover, it was used successfully as chiral auxiliary in aryl–aryl couplings via an SNAr process involving 2-(2-magnesioaryl)oxazolines, the Meyers reaction [22b–d,23]. We decided to introduce the oxazoline group onto the aryl nucleophilic part, that is, the future aryllithium intermediate instead of the aryne, so as to overcome any regioselectivity issues and to better induce atroposelectivity by creating a stereoisomerically well-defined metallacycle by coordination of the oxazoline to lithium. The coordination was expected to take place via the nitrogen atom of the oxazoline in ethereal solvents, as shown by Jantzi et al. [24].
We prepared and assessed various aryloxazolines in the ARYNE coupling. The expected biaryls were formed along with a side product, the corresponding fluorenone resulting from the cyclization of the biaryllithium intermediate, followed by hydrolysis during workup. Considering that the fluorenone byproduct and the desired biaryl are both formed from the same biaryllithium, the total coupling yield has to be taken into account, which ranges from 48 to 79%. Unfortunately, axial chirality is lost in the planar fluorenone, which thus constitutes an issue. Nevertheless, the formed biaryls showed axial stereoenrichment with a narrow 66:34–71:29 range of dr. The diastereoselectivity seems to be slightly influenced by the size of the R2 group (Scheme 9).
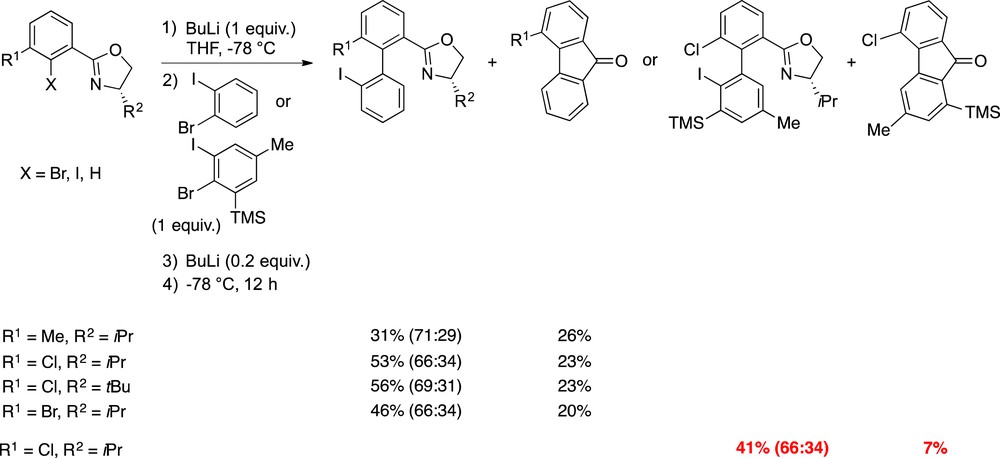
Atropodiastereoselective ARYNE coupling of chiral aryloxazolines with arynes.
The results indicate that with the uncongested aryne precursor a 2.3:1 ratio of biaryl with regard to fluorenone is obtained, whereas the sterically hindered Trimethylsilyl (TMS) analogue afforded a much better 5.9:1 ratio in favor of the desired biaryl. This difference might indicate that cyclization is disturbed by the proximity of the TMS group and the resulting oxazolidine (Fig. 7).

The cyclization issue and the role of bulky substituents in the vicinity of the aryne triple bond.
2.5 Application of the atropodiastereoselective ARYNE coupling to the formal synthesis of (−)-steganacin
On the basis of these findings, we imagined introducing two trimethylsilyl groups on each side of the triple bond of the aryne. The second TMS group shall avoid competitive regioselectivity of the nucleophilic addition onto the aryne. In addition, the versatile silicon–carbon bond can be transformed toward various derivatives. Bis-TMS–substituted benzodioxole-derived aryne precursors were thus prepared and evaluated in the coupling with aryloxazolines. Gratifyingly, the iodinated aryne precursor led to the expected biaryls without any cyclized product with 45–67% yields, which is acceptable for such congested biphenyls (Scheme 10).
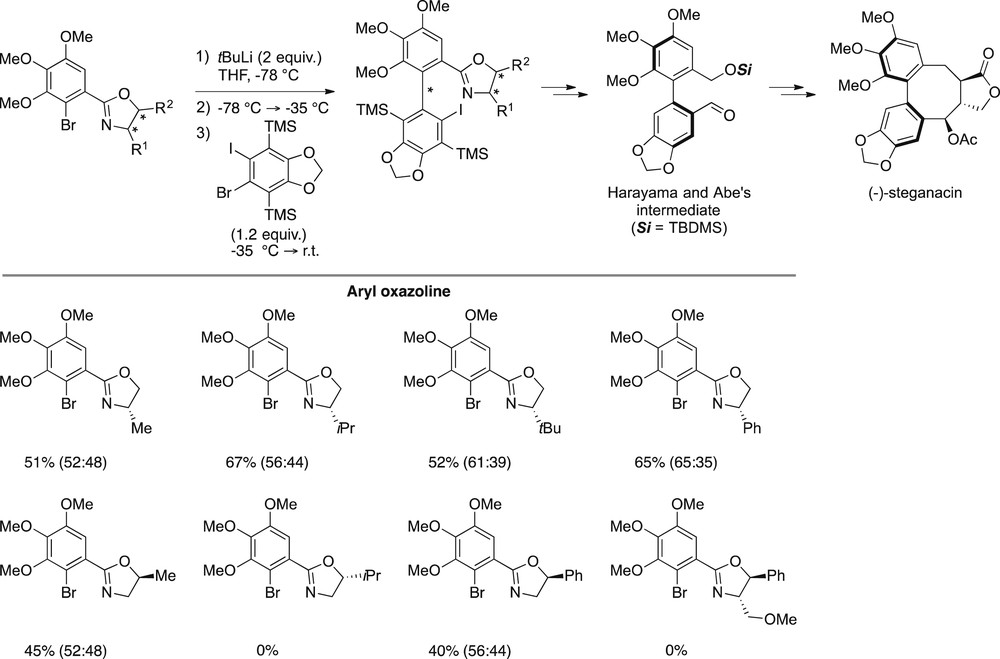
ARYNE coupling of chiral aryloxazolines with a bis-silylated aryne precursor.
Moderate diastereoselectivities were obtained because of presumed opposing substituent effects in competing transition states. Fortunately, atropodiastereomers could be easily separated in several cases. One of them was then successfully converted into Harayama and Abe's key intermediate toward (−)-steganacin after desilylation, hydrolysis of the oxazoline auxiliary into a protected benzylic alcohol, and formylation of the iodine-bearing carbon via iodine/lithium exchange. All of the steps, performed on diastereomerically pure precursors (dr > 98:2) led, without any racemization, to both enantiomers of the target molecule in er > 99:1 [9j,l,25].
3 Conclusion
The “ARYNE” adventure, which started in 2001, led to an efficient major method for accessing ortho, ortho′-di-, tri-, and tetrasubstituted biaryls without any transition metals on multigram quantities. We showed that regioselective bromine/lithium interconversions allow for their functionalization at will. To access enantiopure biphenyls, we developed a desymmetrization/deracemization strategy based on an enantiopure sulfinyl group as chiral auxiliary.
Then, we developed the most challenging atroposelective coupling of in situ generated arynes and aryllithiums bearing various chiral auxiliaries ortho to lithium. tert-Butyl sulfoxides gave low to high yields of the coupling product with variable diastereoselectivity. p-Tolyl sulfoxides, on the other hand, were unable to produce the desired biaryls, mainly because of a lack of reactivity of the 2-lithiophenyl p-tolyl sulfoxide intermediates toward halogen/lithium exchange and/or trapping by the aryne. Tartrate-derived chiral diethers yielded the coupling products with moderate diastereoselectivity. Oxazolines gave the most abundant results in terms of coupling, after an evaluation of different reaction parameters.
Finally, we carried out the synthesis of Harayama and Abe's intermediate of (−)-steganacin in nine steps, 22% overall yield, excellent control of chirality (>98% ee with retention of axial stereoenrichment at each stage of the process), and in the absence of transition metals. Interestingly, the enantiomer of the target compound could be obtained with similar efficiency. The present work enforces the interest in heavy metal-free biaryl synthesis applied to the preparation of potential pharmaceutically relevant ingredients.
Acknowledgments
This work was supported by the French Centre National de la Recherche Scientifique (CNRS) and the University of Strasbourg. The French Agence Nationale pour la Recherche (ANR) (grant number ANR-14-CE06-0003-01, ChirNoCat) is gratefully acknowledged for financial support.