

1 Introduction
In recent years, several chiral ligands and organocatalysts based on helically chiral scaffolds have been described, and some successful applications in enantioselective catalysis have been reported. Recent reviews afford a suitable overview of this field, which include oxygen-, nitrogen-, sulfur-, and phosphorus-functionalized species [1]. This review focuses on the catalytic applications of helicenes bearing phosphorus functions, because significant advances have been obtained in this field during the last 2 years, that were not covered by previous reviews. Most of these advances result from our recent work on the synthesis and uses of phosphahelicenes, whereas new studies on helicenes with appended phosphorus functions have been reported by other groups. Previous catalytic studies and synthetic approaches to phosphorus-functionalized helical derivatives are outlined briefly in this review to give an exhaustive view of the field.
This review is organized in two main sections that deal with distinct families of helical phosphorus derivatives: helicenes with appended phosphorus functions, typified by 1, and phosphahelicenes, typified by 2, respectively (Fig. 1). As shown hereafter, both the synthetic approaches and catalytic uses differ significantly for the two series.
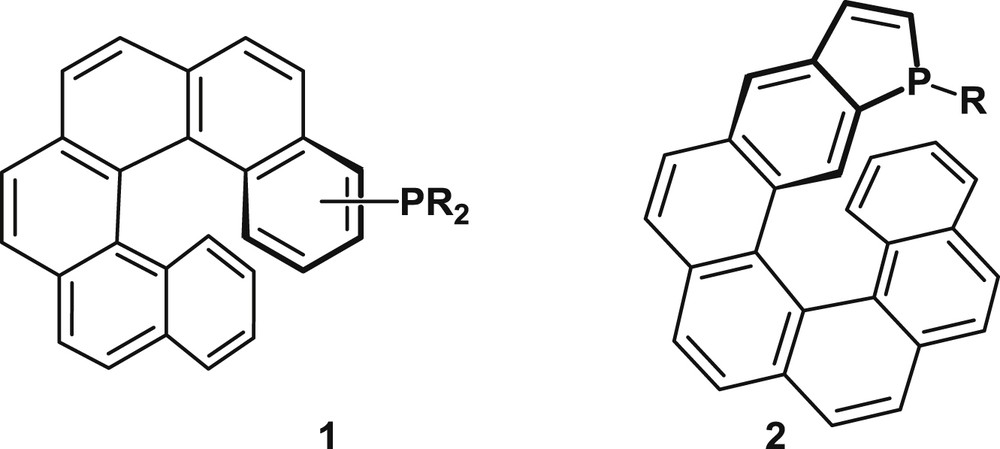
Helical scaffolds with appended phosphorus functions (1) and phosphahelicenes (2).
2 Helicenes with appended phosphorus functions
2.1 Synthesis of helical phosphines
Several helicenes with appended phosphorus functions have been designed with the specific aims of using them as ligands in organometallic catalysis and/or taking advantage of the helical chirality in organocatalytic processes. Representative examples are displayed in Fig. 2.
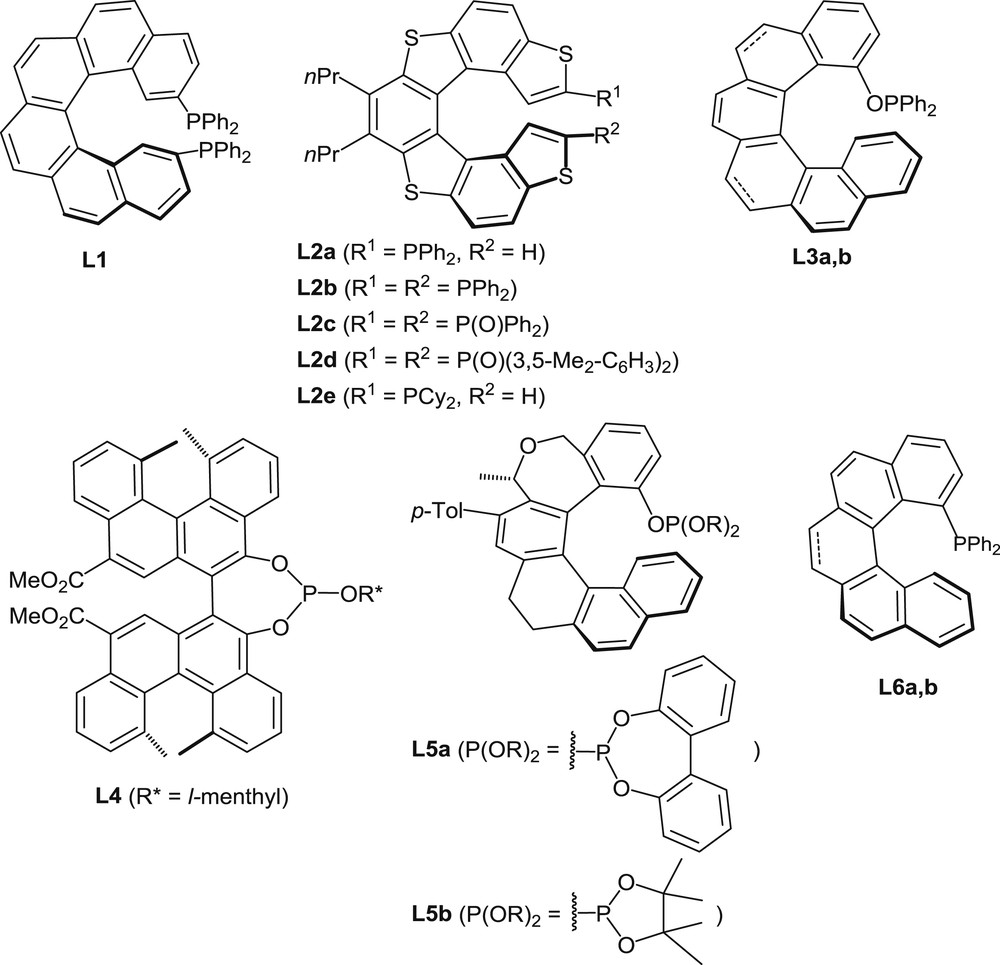
Helicenes with appended phosphorus functions used as ligands in organometallic catalysis or as organocatalysts.
The synthesis of helical phosphines usually involves building of the desired helical scaffold and introduction of the phosphorus functionality at a late stage, a very common procedure being the reaction of a chlorinated phosphorus electrophile with a helical organolithium derivative (see Schemes 1 and 2). Alternatively, a PR2 group could be introduced by palladium-catalyzed coupling of R2PH with bromohelicenes [2]. For the synthesis of phosphites and phosphinites, the usual procedure involves the reaction of helical phenols with chlorophosphanes (e.g., see Schemes 3 and 4). The main strategies to access the corresponding helical backbones rely on either photocyclization of diarylalkene precursors, metal-promoted [2+2+2] cyclization of poly-ynes, or other cycloisomerization reactions. All these methods are typified in sections 2.1.1 to 2.1.3 hereafter by a few representative examples. In addition, helical phosphorus derivatives have been prepared by Katz and co-workers who took advantage of Diels–Alder type reactions on quinones to build highly functionalized helical scaffolds [3]. The resulting phosphine oxides and phosphites have not been used so far in catalysis.
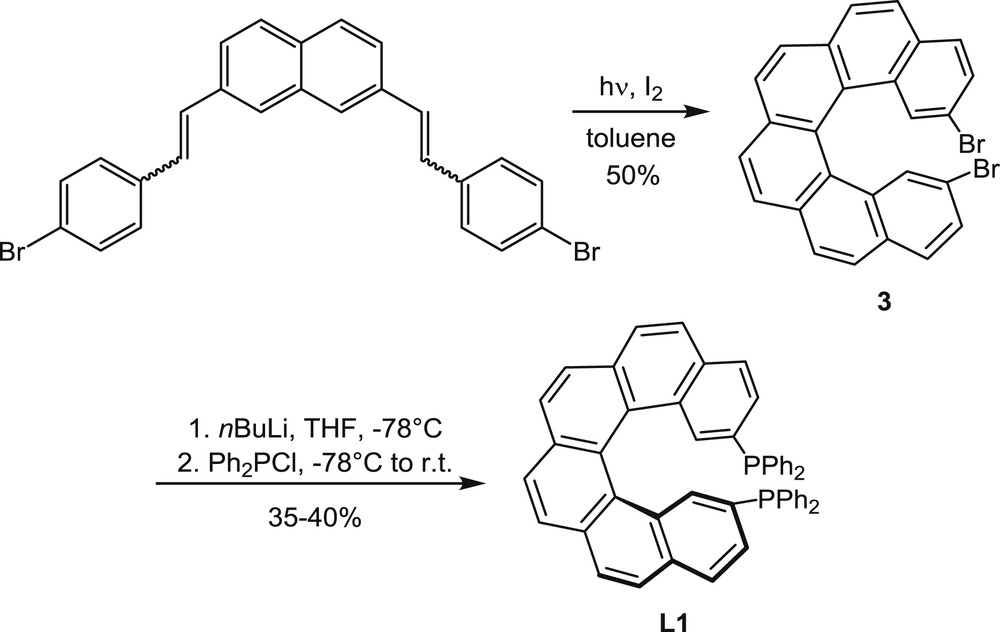
Synthesis of the helical diphosphine L1.
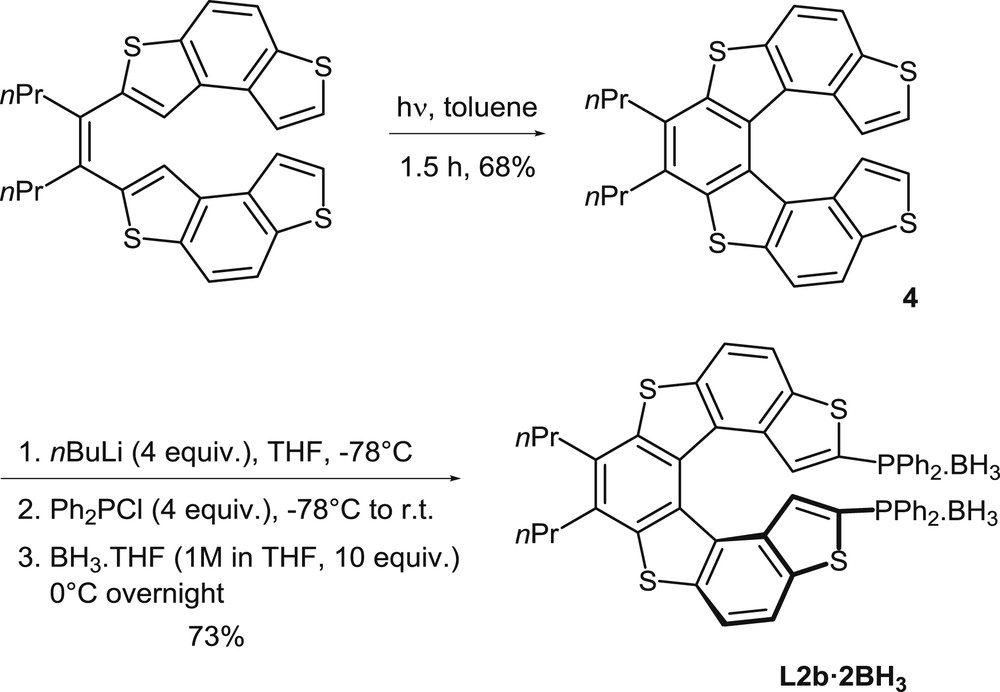
Synthesis of the borane complex of the helical diphosphine L2b.
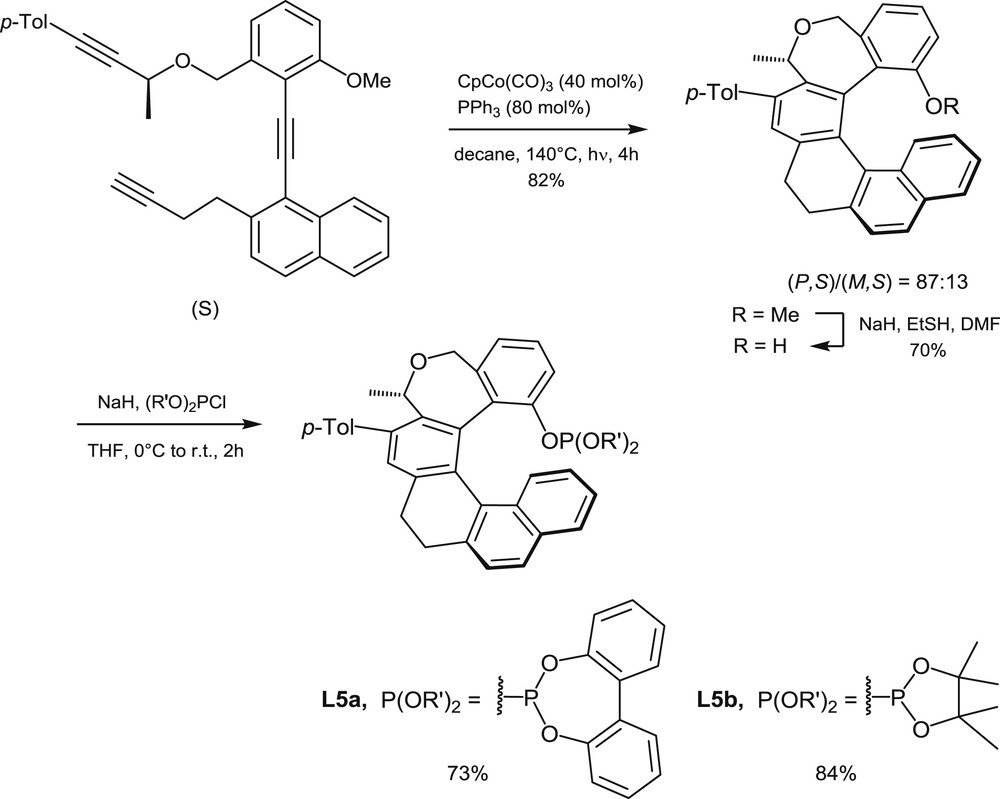
Synthesis of phosphites L5 via diastereoselective cyclotrimerization of triynes.
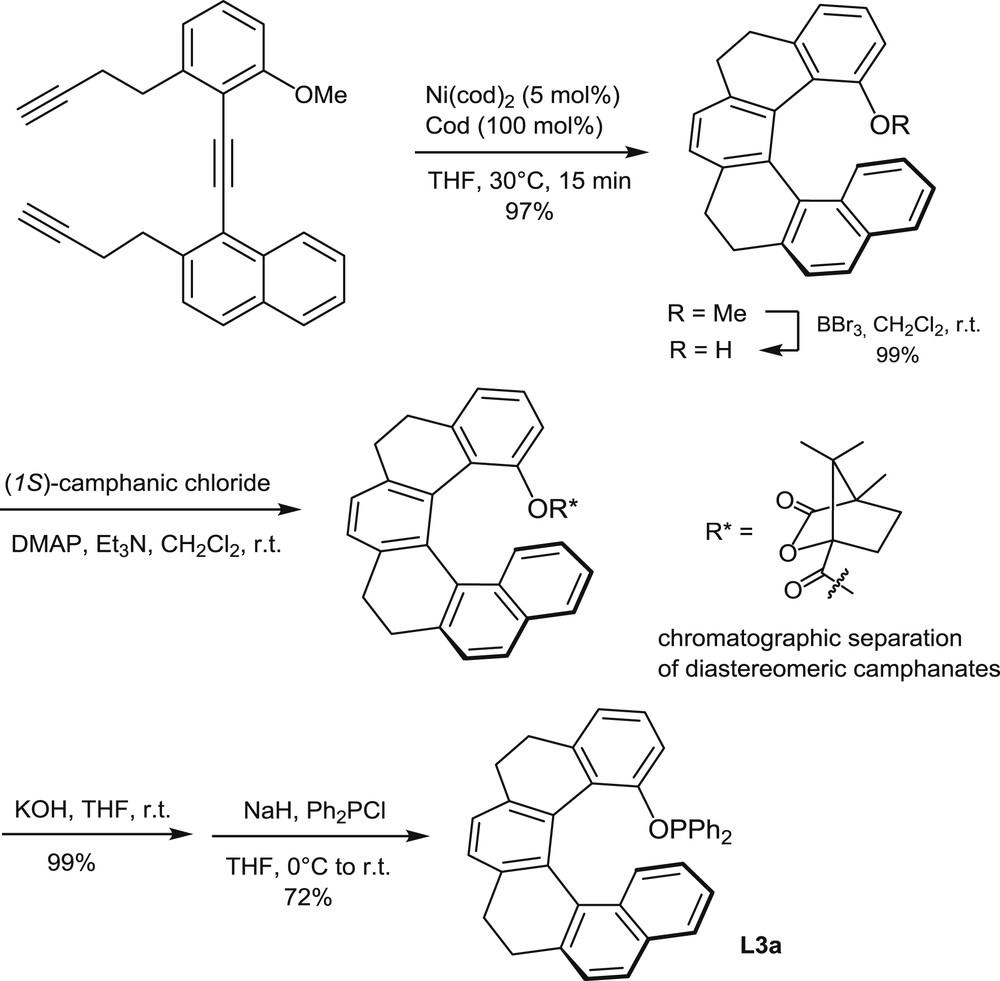
Synthesis of the optically pure phosphinite L3a.
2.1.1 Photochemical approach
The oxidative photocyclization of diaryl olefins is one of the first and most widely used methods for the synthesis of polyaromatic helical compounds [4]. Therefore, this method has been considered in initial studies on the synthesis of helical ligands for catalytic purposes [5] and then largely applied (see examples hereafter). In other studies, helical phosphines obtained by the photochemical approach have been simply characterized, without further uses [6]. The method is illustrated in Scheme 1 by the synthesis of L1 (called Phelix [5]).
2,7-Bis(bromostyryl)naphthalene, which was obtained by Wittig reactions, has been converted into 2,15-dibromohexahelicene 3 by oxidative photocyclization in the presence of iodine as the oxidant. Starting from 3, the diphenylphosphino groups have been introduced via bromine–lithium exchange and the subsequent reaction with chlorodiphenylphosphane. The 2,15-bis(diphenylphosphino)[6]helicene L1 was isolated at first in the racemic form and then in the enantiomerically enriched form, starting from enantioenriched dibromide 3, 96% ee, which was obtained by separation of the racemic mixture by high pressure liquid chromatography (HPLC) using a chiral stationary phase.
The photochemical oxidative cyclization method has been applied also to the synthesis of the tetrathia[7]helicene scaffold of diphosphine L2 (Scheme 2) [7]. The phosphine precursor 4 and other analogues had been prepared by photochemical cyclization of the corresponding olefins, with the initial purpose of investigating their optoelectronic properties [8]. The photocyclization of the starting olefin in toluene takes place under standard conditions and affords 4 in good yield (68% isolated yield).
It has been established that thiahelicene 4 can be easily functionalized then by the direct, selective metalation of the α positions of the terminal thiophene units, by using an excess nBuLi as the metalating agent. The method allows the easy modulation of the substitution pattern, including the introduction of phosphorus substituents, as shown in Scheme 2. Reaction of the dilithiated thia[7]helicene with chlorodiphenylphosphine affords the desired diphosphine L2b, which is converted in situ into its bis-borane complex to facilitate the purification process. Alternatively, the same dilithiated thiahelicene was reacted with Ph2P(O)Cl to obtain the corresponding phosphine oxide [7b]. The borane complex L2b·2BH3 was also prepared in an enantiomerically pure form, starting from its chiral precursor (P)-4, because 4 could be easily separated into pure enantiomers by HPLC [7a]. In a different approach, the enantiomers of the corresponding diphosphine dioxide could be isolated in an enantioenriched form by HPLC separation on a chiral column [7b].
2.1.2 Catalytic cyclotrimerization of triynes
The second strategy to access helical phosphorus derivatives involves building of the helical scaffold by transition metal-promoted intramolecular [2+2+2] cyclotrimerization of triynes [9a]. The cyclotrimerization process allows building of one of the central aromatic rings of the helical sequence, by starting from suitable, polyaromatic triynes. The method has been applied to the synthesis of trivalent phosphines [9b], phosphinites, and phosphites, but only phosphinites L3 [10] and phosphites L5 [9c] have been considered for catalytic applications. The synthesis of phosphites L5, which was reported in 2011 by Starý et al., is shown hereafter to illustrate the catalytic cyclotrimerization method (Scheme 3).
A [2+2+2] cyclotrimerization reaction, promoted by a CpCo(CO)3/PPh3 1:2 mixture, has been carried out on the optically pure triyne displayed in Scheme 3 [9d]. Under thermodynamic conditions, that is, under heating at 140 °C, the reaction led to an 87:13 mixture of two diastereomeric oxa-helicene derivatives (R = OMe) with opposite configurations of their helical scaffolds (82% yield). Then the pure (P,S) diastereomer was isolated in 63% yield by crystallization and proved to be configurationally stable at room temperature. The corresponding helical phenol (R = H) served then as a starting material for the syntheses of phosphites L5a and L5b, which were carried out by deprotonation of the phenol with NaH and subsequent addition of the corresponding chlorophosphites.
An analogous cyclotrimerization approach has been used recently for the preparation of the [6]helicene-based phosphinites L3a and L3b [10]. The intramolecular cyclotrimerization of the triyne shown in Scheme 4 has been performed on a multigram scale (>10 g), with Ni(cod)2 as the catalyst (5 mol %), in the presence of an excess of cod (cod = 1,8-cyclooctadiene), at 30°C. After conversion of the helical ether (R = OMe) into the corresponding phenol, the racemic phenol was resolved into enantiomers by separation of the diastereomeric (1S)-camphanates. Then the phosphinite function was created by a reaction of the optically pure phenol with diphenylchlorophosphine in the presence of NaH. The phosphinite L3a was obtained in 72% yield.
Alternatively, the partially saturated helical phenol in Scheme 4 was dehydrogenated with Ph3CBF4 to afford a fully aromatic helicene and the corresponding phosphinite L3b through the same reaction sequence [10].
2.1.3 Platinum(II)-catalyzed cycloisomerization
Very recently, an alternative synthetic approach to the helical scaffolds of chiral phosphines has been described by Usui [11a]. It involves a PtCl2-promoted cycloisomerization as the key step, as shown in Scheme 5 [11b]. This reaction affords a partially saturated 1-bromo[5]helicene, which serves as a starting material for the introduction of the diphenylphosphino group through the usual metalation/phosphination procedure. In this case, resolution has been performed on the phosphine oxide by chromatographic separation and crystallization of its chiral spiro-TADDOL adducts (TADDOL = α,α,α′,α′-tetraphenyl-1,3-dioxolan-4,5-dimethanol).
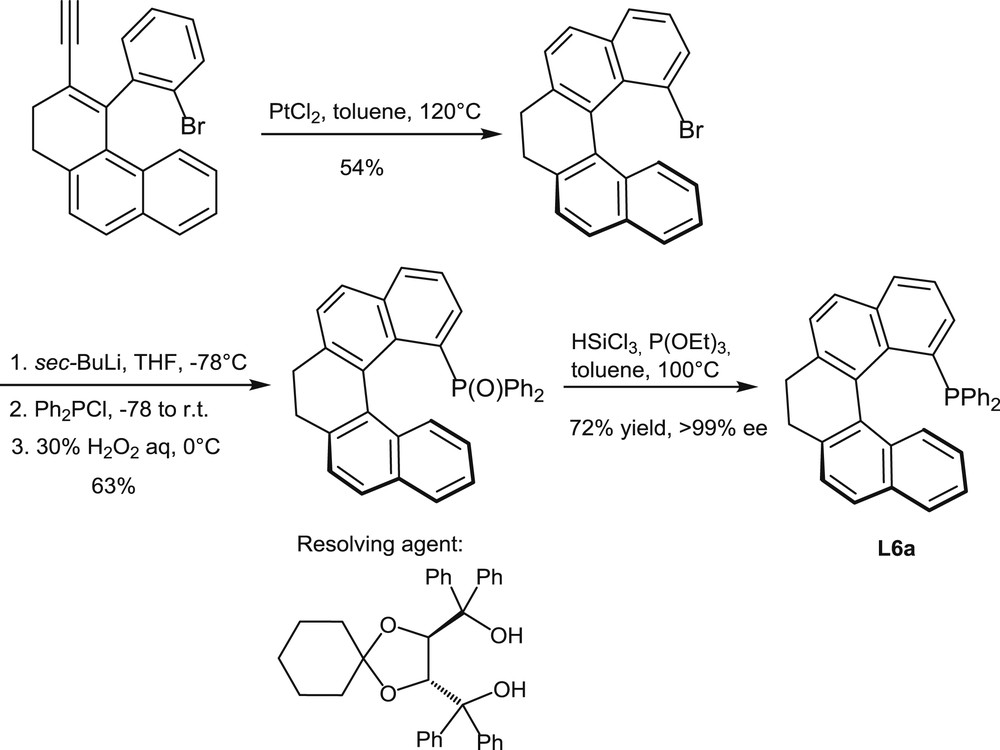
Synthesis and resolution of the helical monophosphine L6a.
The same reaction sequence affords the analogous, fully aromatic helical phosphine L6b when the enantiomerically pure phosphine oxide is dehydrogenated with DDQ before reduction (DDQ = 2,3-dichloro-5,6-dicyano-1,4-benzoquinone).
2.2 Catalytic applications
The following section summarizes the known applications of helicenes L1–L6 in organocatalysis and organometallic catalysis, which include palladium, iridium, and rhodium catalysis, with special focus on asymmetric processes. In addition, preliminary results are also reported on the catalytic uses of gold complexes of racemic helical phosphines L2.
2.2.1 Screening in palladium-catalyzed asymmetric allylic alkylation
In 2000, Reetz and Sostmann [12] have reported on the first uses of helical phosphines in palladium catalysis, by disclosing that palladium complexes of phosphine L1 are able to promote enantioselective allylic substitutions. The study focused on the reaction of the standard substrate, 1,3-diphenylpropenyl acetate with dimethyl malonate, leading to 6 (Scheme 6).
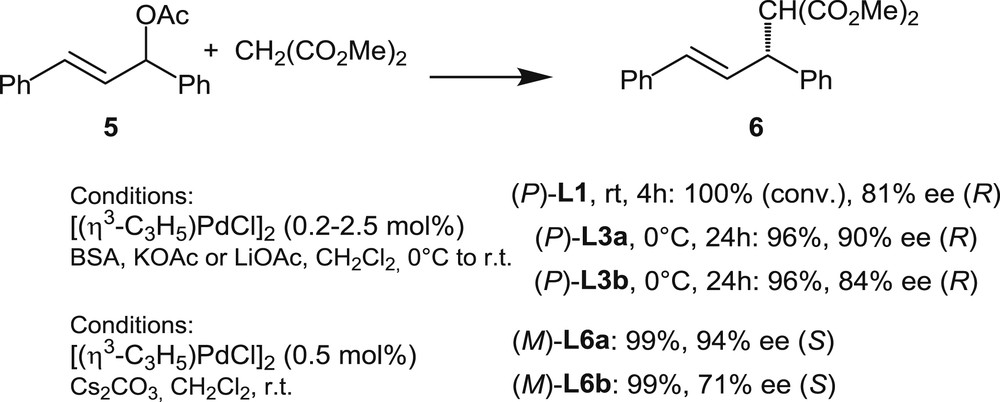
Helical ligands L1, L3, and L6 in Pd-catalyzed asymmetric allylic alkylations with dimethyl malonate.
The authors demonstrated that an L1/Pd 2:1 ratio is optimal to efficiently catalyze the reaction. Total conversion was obtained after 4 h, and the desired product was isolated in 81% ee. 31P NMR studies of the catalytic mixture [(η3-C3H5)PdCl]2/L1 showed that various species are present in solution. Moreover, the ligand/metal ratio did not seem to have a significant effect on the enantioselectivity of the reaction. These observations founded the assumption that L1 behaves as a monodentate ligand. In the same article, the authors also reported on the kinetic resolution of 1,3-diphenylpropenyl acetate 5. Starting from racemic 5, analysis of different aliquots showed that the enantiomeric excess of the product 6 remains constant over the reaction. However, the enantiomeric excess of the remaining substrate was found to increase during the catalytic reaction, reaching >97% ee at about 60% conversion. A similar behavior was observed in the analogous allylic substitution on 1,3-diphenylallyl benzoate.
In 2016, Tsujihara et al. [10] reported on the first examples of allylic alkylations promoted by palladium complexes of helically chiral phosphinites. Phosphinites L3a,b have been tested in the model allylic alkylation shown in Scheme 6. The catalysts were formed in situ from L3 and [Pd(η3-C3H5)Cl]2 under conditions very similar to those previously applied by Reetz and Sostmann, but the ligand to Pd ratios were increased to 4:1. The desired product 6 was isolated in high yield (96%) and 90% ee when L3a was used, whereas the fully aromatic helicene L3b proved slightly less effective as it afforded the desired product with only 84% ee. These recent results demonstrate the potential utility of L3 as chiral ligands in asymmetric catalysis.
Finally, the same reactions have been carried out with palladium catalysts based on phosphines L6a,b (1:1 phosphine/palladium ratio) [11a]. The same trend as with L3a,b has been noticed: excellent yield and enantiomeric excess have been obtained with the partially saturated [5]helicene L6a, whereas the fully aromatic analogue L6b gives a lower enantiomeric excess, 71% ee.
The excellent behavior of L6a in these model reactions encouraged more extended investigations on palladium-promoted allylic nucleophilic substitutions. Indoles and alcohols have been considered as nucleophiles in reactions with 1,3-diphenylpropenyl acetate in the presence of the Pd(II)/L6a catalyst. Excellent yields and enantiomeric excesses have been obtained as summarized in Scheme 7 [11a].
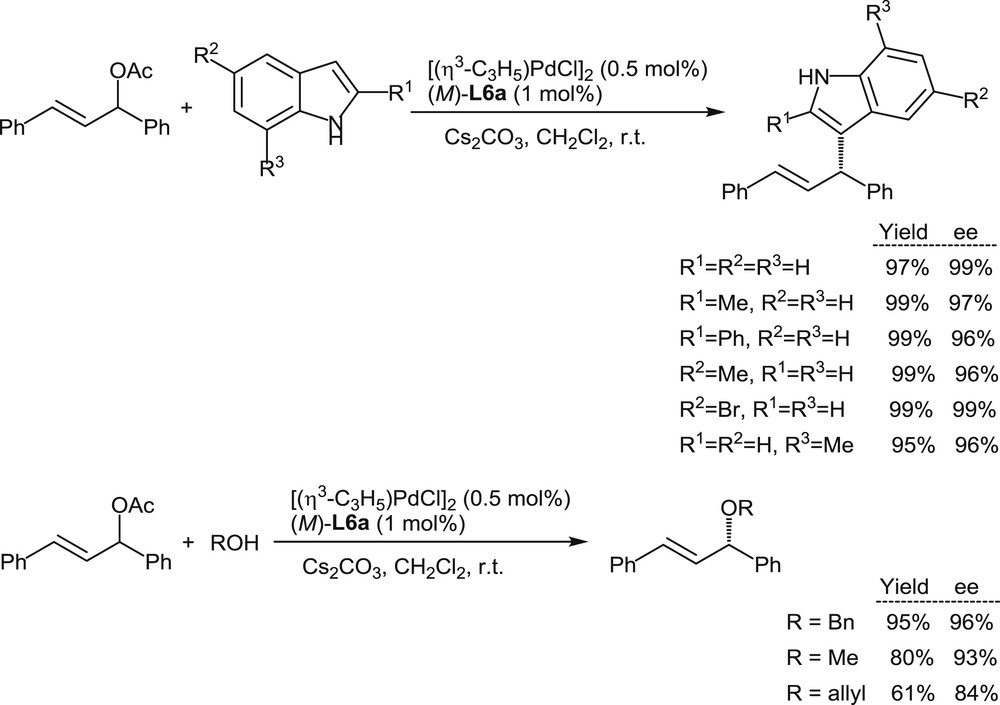
Palladium-promoted substitutions on diphenylpropenyl acetate with indoles and alcohols as the nucleophiles.
Especially noteworthy are the high enantioselectivity levels attained in the reactions with indole nucleophiles, which with other catalysts met so far with only limited success.
2.2.2 Screening in palladium-catalyzed asymmetric Suzuki–Miyaura coupling reactions
Encouraged by the high enantioselectivity levels attained in the allylic substitution reactions mentioned previously (Schemes 6 and 7), the use of [5]helicenes L6a,b as chiral ligands has been extended to Suzuki–Miyaura couplings of 1-bromo-2-naphthylphosphonates with arylboronic acids (Scheme 8) [11a]. In this case, the fully aromatic ligand (P)-L6b outperforms its analogue (P)-L6a and affords good yields and high enantiomeric excesses for a series of variously substituted substrates (nine examples, 83–99% ee).
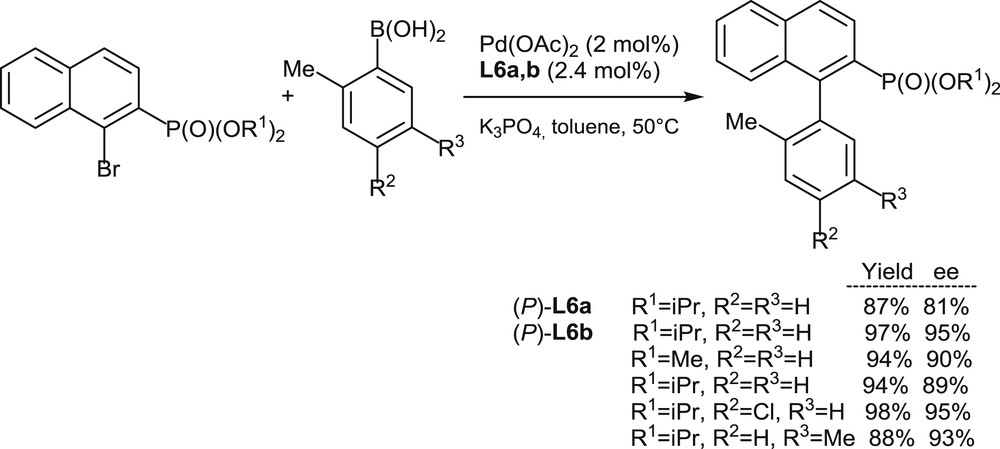
Palladium-promoted enantioselective Suzuki–Miyaura couplings.
Overall, the recent studies on ligands L6a,b (Schemes 7 and 8) [11a] fully demonstrate, for the first time, the high potential of helical phosphines in challenging enantioselective palladium-promoted reactions. A key point in the design of these phosphines seems to be the positioning of the phosphorus substituent on the internal edge of the helical structure at its most hindered 1-position. Such structural feature ensures both the configurational stability of the [5]helicene scaffold and a highly asymmetric, highly discriminating environment to the metal center in the corresponding complexes.
2.2.3 Screening in rhodium-promoted hydrogenations and hydroformylations
Several chiral helicenes have been investigated as ligands in the rhodium-promoted hydrogenation of functionalized olefin. The first example, described by Reetz et al. [5], was dedicated to the hydrogenation of the itaconic acid ester 7 with a preformed catalyst, which was prepared from the [6]helicene-based diphosphine (P)-L1 and a stoichiometric amount of (cod)2RhBF4 (Scheme 9). The reaction was performed at room temperature using dichloromethane as the solvent, at a H2 pressure of 1 atm, with a 0.1 mol % catalyst loading. The desired product 8 was obtained in 54% yield with a moderate 39% ee in favor of the S-enantiomer.
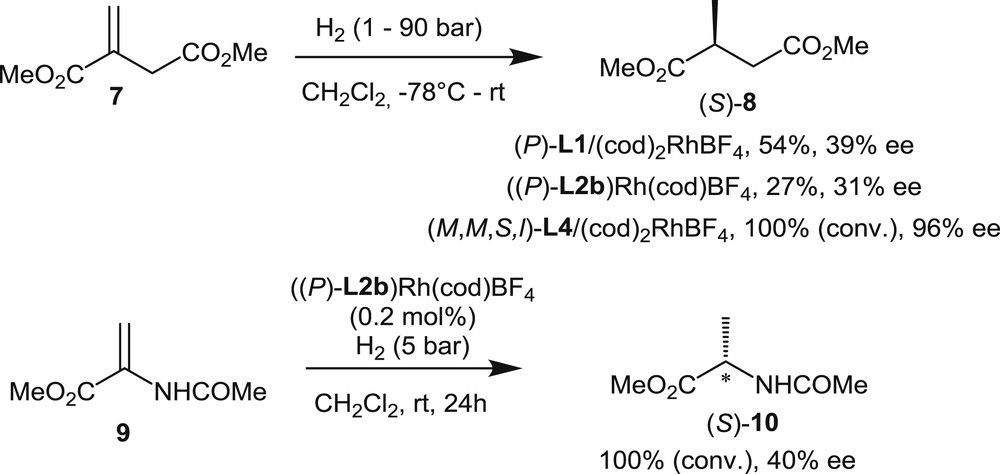
Enantioselective hydrogenations of dimethyl itaconate and 2-acetamidoacrylate with rhodium complexes of helical phosphines and phosphites.
Later on, the hydrogenation of both itaconic ester 7 and methyl 2-acetamidoacrylate 9 was investigated by Licandro et al. [7a] using a preformed cationic rhodium complex of the optically pure thiahelicene-derived diphosphine (P)-L2b [7a]. The reactions were performed at room temperature under a 5 bar H2 pressure. In both cases, the enantiomeric excesses of the products were low (31% ee and 40% ee, respectively).
Finally, the most successful hydrogenation of 7 was reported by Nakano and Yamaguchi [13]. Rhodium complexes were formed from the four distinct diastereomers of phosphite L4 (Fig. 2). The diastereomers differ by the relative configurations of their chiral units: the helical chirality of the [4]helicene fragment, the axial chirality of the binaphthyl unit, and the central chirality of the l-menthyl substituent on phosphorus. Screening of the four preformed catalysts in the hydrogenation of dimethyl itaconate 7 at 90 atm H2 pressure revealed that phosphite (M,M,S,l)-L4 gives the highest enantiomeric excess (96% ee, Scheme 9). The authors demonstrated that the “match/mismatch” effect between the helical and axial chirality plays a crucial role in asymmetric induction, whereas the configuration of the menthyl group does not seem to have a significant influence. These studies also showed that H2 pressure strongly affects the enantioselectivity levels. Indeed, when the H2 pressure was decreased to 40 atm, the enantiomeric excess decreased to 77%. Although these phosphite-based rhodium catalysts perform better than helical diphosphine rhodium complexes, these catalysts remains noncompetitive with other known systems, in terms of both catalytic activity and enantioselectivity.
In 2011, the asymmetric rhodium-catalyzed hydroformylations of styrene, p-chlorostyrene, and vinyl acetate were investigated by Starý et al. using the helical phosphites L5 [9c]. Phosphites L5 display the same, partially saturated helical backbone and different dioxaphosphepine units connected to the most hindered C1-position of the helicene.
In the hydroformylation of styrene (Scheme 10), the best catalytic system was obtained by combining Rh(acac)(CO)2 as the rhodium source and phosphite L5a, which displays a seven-member cyclic phosphite unit derived from (1,1′-biphenyl)-2,2′-diol. It operates at 50 °C in toluene, under CO (10 bar) and H2 (10 bar), with a 1% catalyst loading, giving 96% conversion rate after 20 h. Although the selectivity toward the branched product 12 over the linear one, 13, is satisfying, the enantiomeric excess remains very low (29% ee).
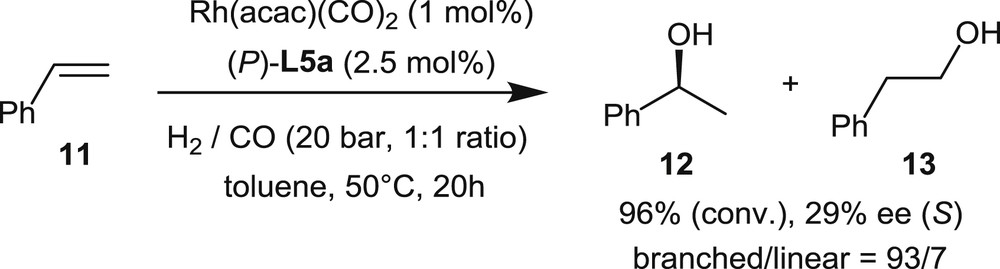
Rhodium-promoted hydroformylation of styrene.
The same low levels of enantioselectivity (<20–25% ee) have been obtained in the hydroformylation of p-chlorostyrene and vinyl acetate.
2.2.4 Screening in enantioselective iridium-promoted allylic aminations
One of the most successful applications of helicenes with appended phosphorus functions is the iridium-catalyzed amination of allylic carbonates. Four phosphites were screened by Starý et al. [9c] in the amination of cinnamyl methyl carbonate 14a and (pyridinyl)allyl methyl carbonate 14b with primary and secondary amines (benzylamine, pyrrolidine, morpholine, and piperidine). The best results were obtained with phosphite L5b, which comprises a tetramethyl-1,3,2-dioxaphospholane unit. Selected examples are shown in Table 1.
Allylic aminations promoted by iridium complexes of the helical phosphites L5b.
| Substrate | Amine | Conversion (%) | Branched/linear | ee (%) |
| 14a | PhCH2NH2 | 95 | >97:3 | 90 (S) |
| 14a | Pyrrolidine | 60 | >99:1 | 92 (+) |
| 14a | Morpholine | 69 | >95:5 | 91 (+) |
| 14b | Piperidine | 70 | >99:1 | 94 (+) |
The catalysts were formed in situ by combining [Ir(COD)Cl]2 and two equivalents of L5b. The reactions were performed in CH2Cl2 (THF gives lower conversion rates) at 35 °C. They gave excellent regioselectivity, in favor of the branched product, and enantiomeric excesses higher than 90% for the four examples reported so far. These encouraging results highlight once more the potential of helical ligands in asymmetric organometallic catalysis.
2.2.5 Screening in gold-promoted cycloisomerization reactions
In a very preliminary study, gold complexes of the racemic helical mono- and di-phosphines L2a and L2b have been prepared by Licandro et al. [14]. These complexes have demonstrated a satisfying catalytic activity in selected cycloisomerization processes involving allenes. Representative examples are the intramolecular hydroarylation and hydroxycarbonylation of allenes shown in Scheme 11.
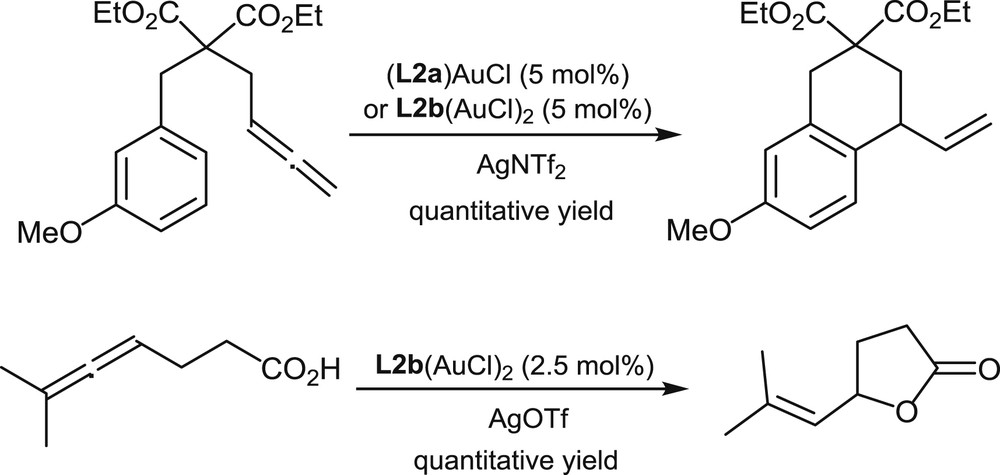
Gold-promoted hydroarylation and hydroxycarbonylation of allenes.
These reactions were conducted at room temperature and monitored in situ by NMR. The article does not mention studies on the asymmetric variants of these reactions.
2.2.6 Organocatalysis with helical phosphine oxides and trivalent phosphines
The first report on the use of helicenes with appended phosphorus functions in organocatalysis has been published in 2011 by Cauteruccio et al. [7b]. The helical diphosphine dioxides L2c,d, based on a tetrathia[7]helicene scaffold, have been tested as Lewis base catalysts in three reactions involving activation of silicon reagents: the aldol reaction, the imine reduction, and the reductive aldol reaction shown in Scheme 12. The yields of these reactions were low to good (40–87%), but the enantiomeric excesses were unsatisfying, with a maximum 22–23% ee. Nonetheless, these preliminary results have represented interesting starting points, which introduced the use of helical chirality in asymmetric organocatalysis.
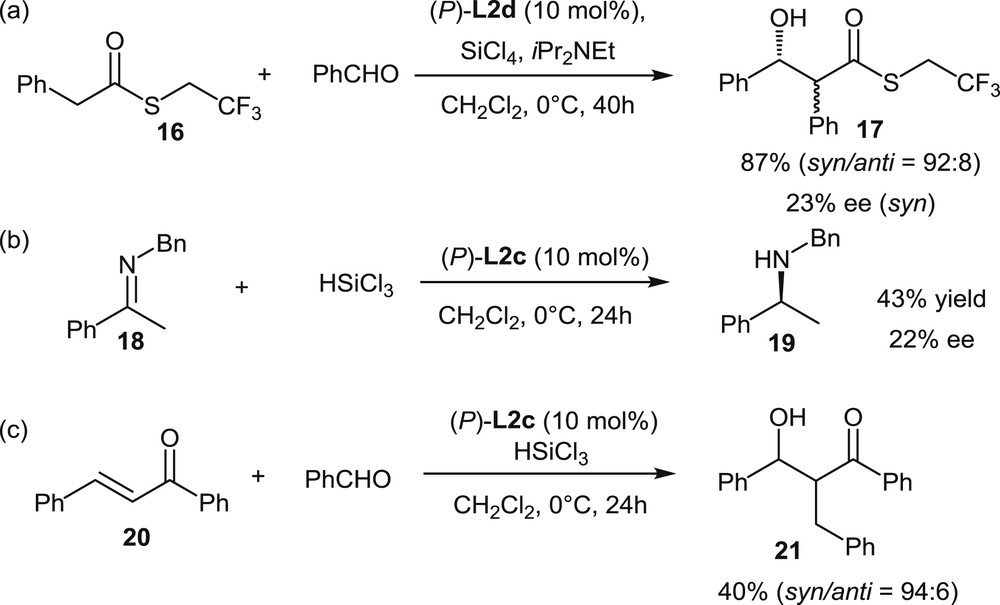
Screening of helical phosphine oxides as Lewis base organocatalysts.
Cauteruccio et al. [7b] have also disclosed the use of the P-dicyclohexylphosphine L2e in [3+2] annulation reactions between allenoates and olefins (Scheme 13) [15]. In these reactions, the trivalent monophosphine behaves as a nucleophilic catalyst through activation of the electron-poor allene [16]. The reactions have been performed at room temperature with 20 mol % of racemic L2e in toluene, affording the desired products in good yields.
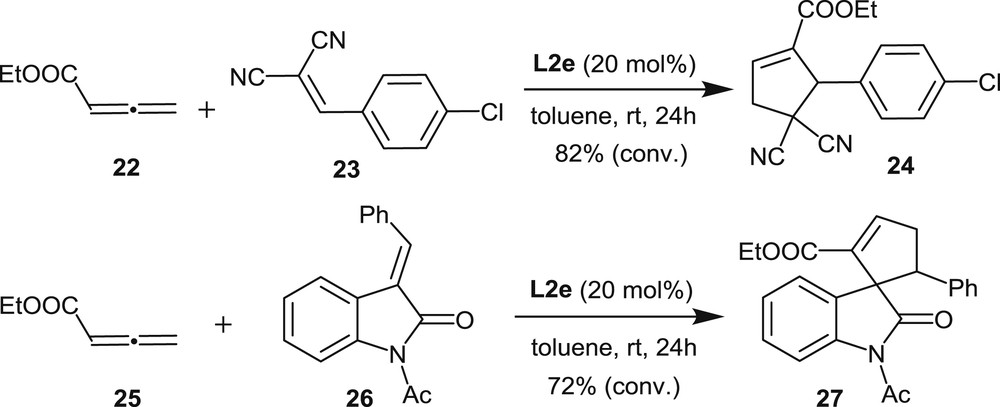
[3+2] Annulation reactions promoted by the trivalent phosphine L2e.
Enantioselective variants of these reactions have not been reported so far. Thus, generally speaking, the chemistry of both phosphine oxides and trivalent helical phosphines of this class as nucleophilic organocatalysts is still at a very early stage of development. Good catalytic activity has been observed, but the enantioselectivity levels remain low in the few reactions considered so far. More promising results have been obtained in this field with the second class of helical phosphines described hereafter (see Section 3.2.2).
3 Phosphahelicenes
Phosphahelicenes are helical compounds in which a phosphorus-containing ring is included into the helical sequence of fused aromatic units. So far only phosphole rings have been used for building such scaffolds. Representative synthetic approaches and applications of these compounds in gold catalysis and nucleophilic organocatalysis are summarized hereafter.
3.1 Approaches for the synthesis of phosphahelicenes
All of the phosphahelicenes used so far in catalysis have been prepared starting from suitably functionalized phosphindoles, such as 28 in Scheme 14, as the key building blocks. These phosphorus-containing units have been incorporated into polyaromatic unsaturated substrates, which have been converted into helical derivatives by following strategies previously applied to the synthesis of carbohelicenes and heterohelicenes, other than phosphahelicenes. These strategies are the photochemical cyclization–dehydrogenation of diaryl olefins and the metal-promoted cyclotrimerization of alkynes. For completeness, it must be noted here that other approaches to phosphahelicenes have been reported, which involve building of a phosphole unit as the final step of the reaction sequence [17]. The resulting phosphahelicenes have not been used in catalysis.
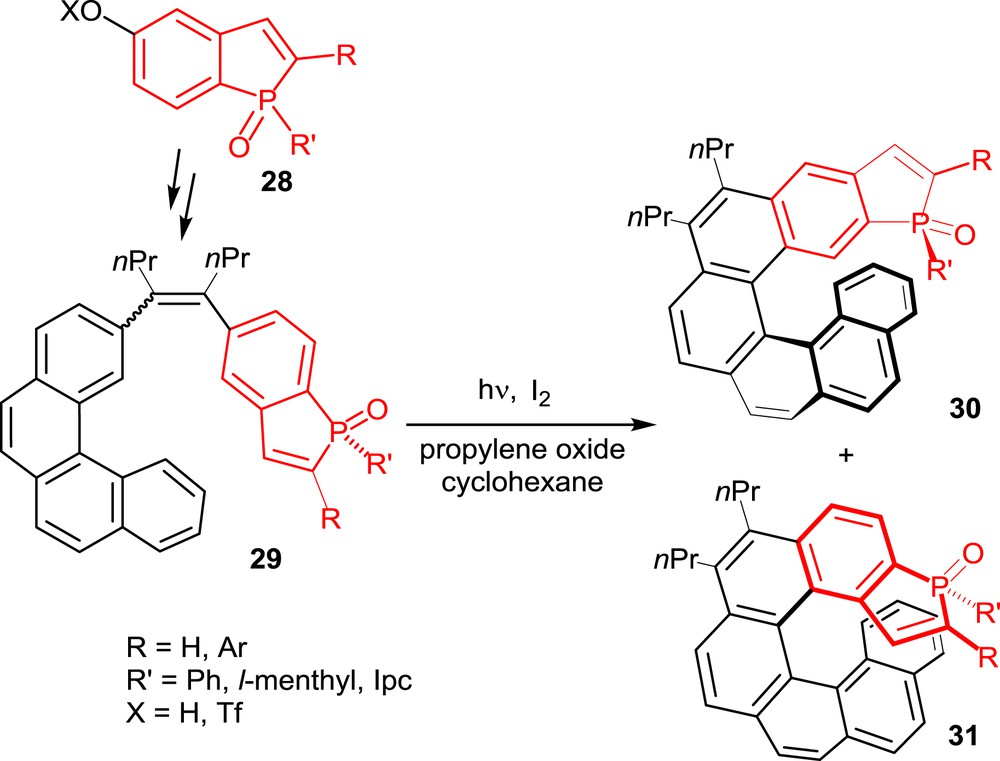
Synthesis of phosphahelicenes oxides via the photochemical approach.
3.1.1 Photocyclization
The oxidative photocyclization of diaryl olefins [4] has been applied to the synthesis of phosphahelicenes by following the general strategy typified in Scheme 14. It consists in the synthesis of a diaryl olefin, such as 29, including a phosphindole oxide unit, which is then submitted to photocyclization under oxidative conditions, with I2 as the oxidant and propylene oxide as the HI scavenger [18].
The photocyclization step usually affords two helical compounds, 30 and 31, depending on the carbon atom involved in the cyclization process, with moderate to good regioselectivity. These reactions are, however, totally diastereoselective in compounds 30 and 31, the phosphorus substituent R′ is selectively oriented toward the helical scaffold and the P(O) function on the external face.
By taking advantage of the high diastereoselectivity of this method, enantiomerically pure phosphahelicenes could be prepared starting from single epimers of the phosphindole oxides, which bear chiral phosphorus substituents, that is, l-menthyl (32a,a′) [18a] or isopinocampheyl (32b,b′) [19] substituents (Fig. 3). Pure epimers of the phosphindoles are isolated easily by column chromatography. In the photocyclization step, the stereochemistry of the phosphorus center dictates the sense of the helical chirality, leading to single epimers of the corresponding helicenes.
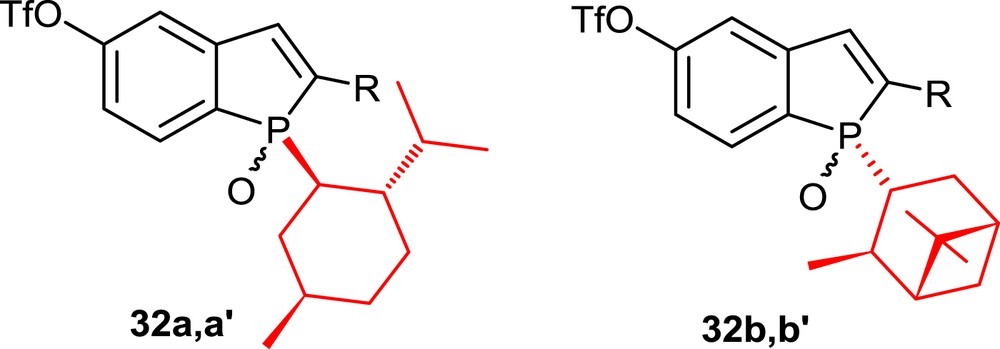
P-l-menthyl-substituted and P-isopinocampheyl-substituted phosphindole oxides.
The method has been applied to the synthesis of a variety of phosphahelicenes, including [6], [7], and [8]helicenes, phosphathiahelicenes, helicenes with unsubstituted and substituted phosphole units (R = H or Ar), in either racemic or enantiopure forms.
In the phosphathiahelicene series (30b), additional modulations of the substitution patterns have been performed by regioselective bromination/palladium-promoted couplings on the terminal thiophene unit of the preformed helicene (Scheme 15) [20].
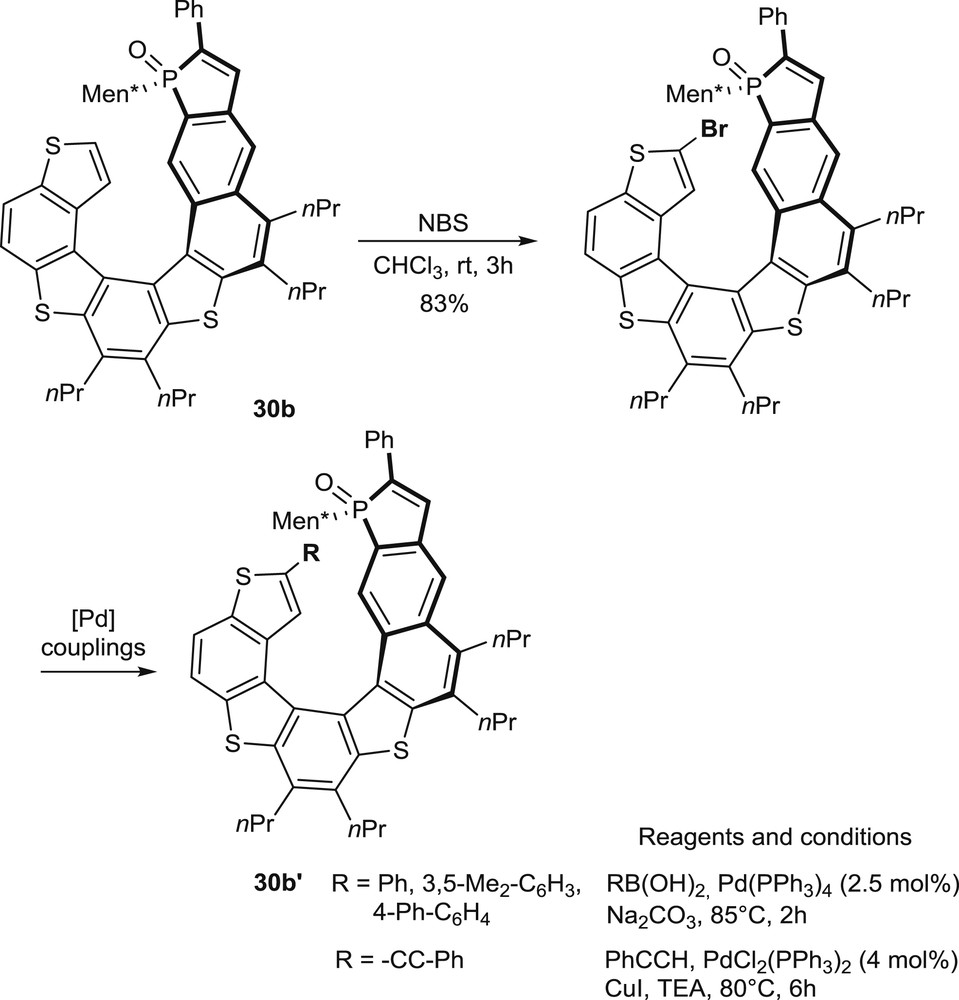
Regioselective bromination on the preformed phosphathiahelicene oxide 30b and subsequent couplings.
This late functionalization approach facilitates the modulation of the substitution pattern and, therefore, the design and synthesis of optimized ligands and catalysts.
3.1.2 Nickel(0)-promoted [2+2+2] cyclotrimerization of triynes
The second synthetic approach involves the intramolecular [2+2+2] cyclizations of triynes as the key step. To obtain phosphahelicenes, two of the known cyclotrimerization strategies described by Stará and co-workers [21] have been applied to triynes, which include phosphindole oxide units in their structures [22]. The [2+2+2] cyclotrimerizations of these substrates are suitably promoted by Ni(cod)2 in the presence of PPh3. A representative example is displayed in Scheme 16 [22b].
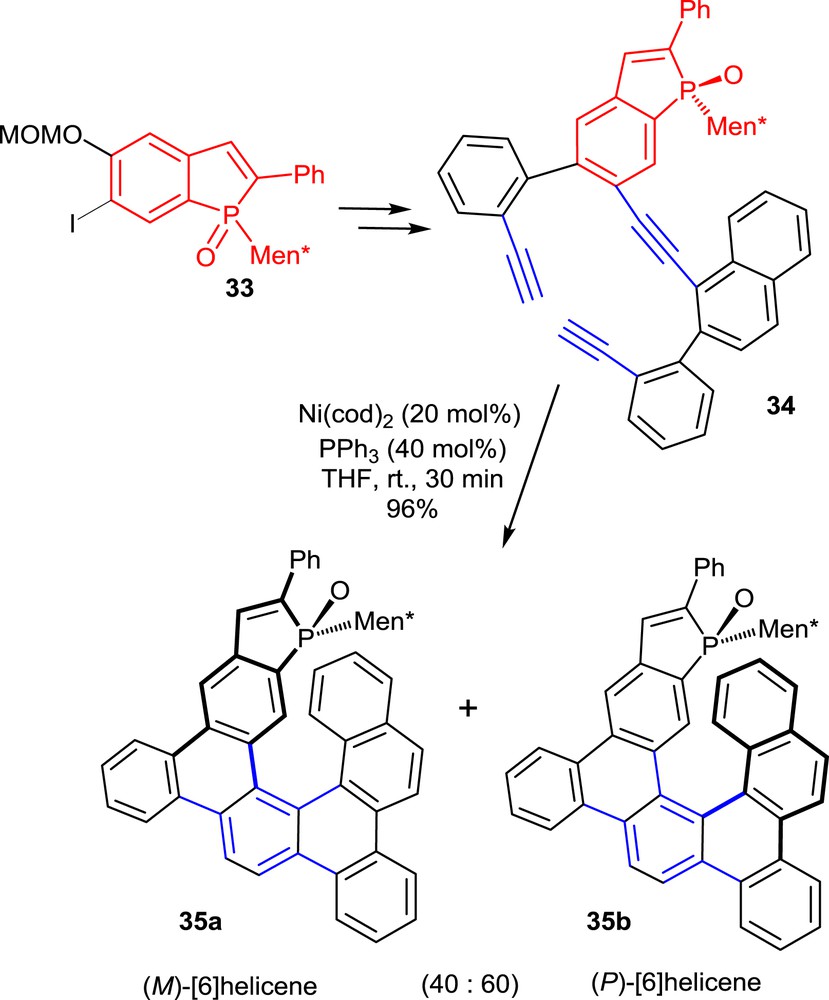
Synthesis of phosphahelicenes via Ni-promoted cyclotrimerization of triynes.
The required triyne 34 was obtained from the phosphindole oxide 33 in three main steps including palladium-promoted Sonogashira and Suzuki couplings with a naphthyl-substituted terminal alkyne and a suitably functionalized aryl boronate, respectively.
Unlike the photocyclization reaction mentioned in the previous paragraph, the nickel(0)-promoted cyclotrimerizations did not display high diastereoselectivity. Therefore, mixtures of the two diastereomeric phosphahelicene oxides 35a and 35b with opposite configurations of the helical scaffolds were obtained in about 4:6 ratio. For catalytic purposes, the epimeric phosphahelicenes 35a,b have been separated by chromatography, so as to obtain enantiomerically pure ligands.
3.2 Catalytic applications
So far phosphahelicenes have been investigated as both chiral ligands in gold-catalyzed cycloisomerization reactions and chiral organocatalysts in [3+2] cyclization reactions. These ligands/organocatalysts are single epimers of phosphahelicenes bearing either menthyl or isopinocampheyl substituents on phosphorus as the chiral auxiliaries. The targeted fields, that is, enantioselective gold catalysis [23] and phosphine organocatalysis [16b,24] have been highly active research domains during the last 10 or 15 years, nevertheless the obtainment of high enantioselectivity levels and the design of wide-scope catalysts still remain challenging. The results obtained with phosphahelicenes in these fields compare favorably with previous literature data and open new promising perspectives.
3.2.1 Gold catalysis
3.2.1.1 Phosphahelicene–gold complexes
The use of phosphahelicenes in gold catalysis has required at first the synthesis and characterization of the corresponding gold complexes, after reduction of the phosphine oxides into trivalent phosphines [18a,b,20,22b,25]. A representative example of the synthesis of phosphahelicene–gold complexes is displayed hereafter in Scheme 17 [25].
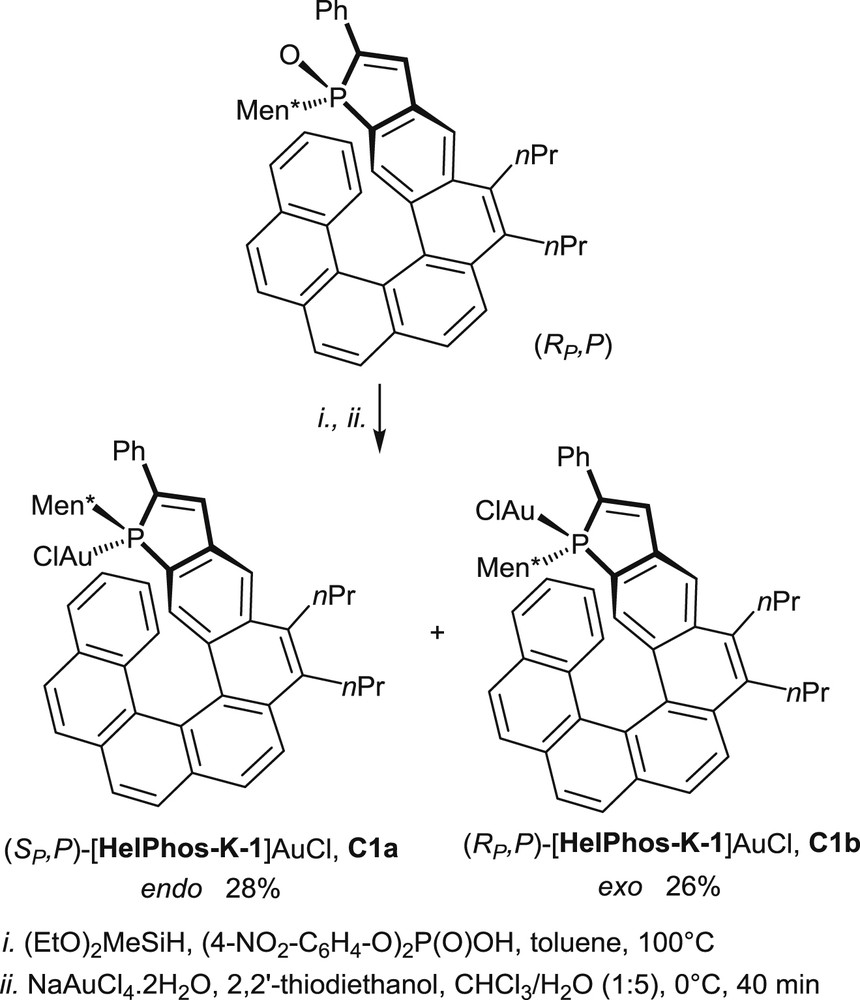
Synthesis of the gold complexes (SP,P)C1a and (RP,P)C1b.
Reductions of the enantiopure phosphine oxides are carried out on single diastereomers, mostly with a (EtO)2MeSiH/(4-(NO2)C6H4O)2P(O)OH mixture at 100 °C [26], but, alternatively, HSiCl3/Et3N at room temperature [18b] or PhSiH3/(4-(NO2)C6H4O)2P(O)OH at 100 °C [20,25] can be used as reducing agents.
The corresponding trivalent phosphines are formed as mixtures of two equilibrating epimers, irrespective of the reduction method. Indeed, in these compounds, the phosphorus center is known to racemize at room temperature, whereas the helical scaffold retains its configuration. The crude mixtures are converted into the epimeric gold complexes by a reaction with either [(HOCH2CH2)2S]AuCl in CHCl3/H2O at 0 °C or the commercially available (Me2S)AuCl complex. In the resulting Au(I) complexes the gold atom may be oriented toward the helical scaffold (endo-complex) or located on the opposite face (exo-complex).
X-ray diffraction studies have allowed accurate structural assignments, and notably assignments of the endo/exo configurations to these complexes. As an example, the crystal structure of the endo-complex (SP,P)-[Helphos-K-1]AuCl, C1a, is displayed in Fig. 4 [25].
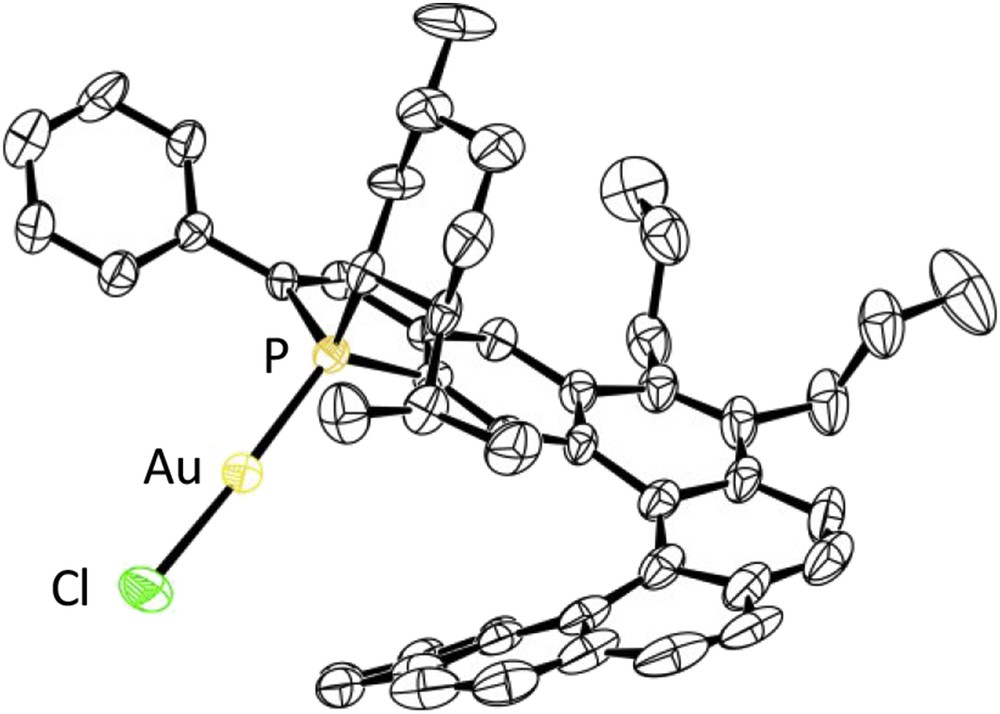
ORTEP drawing of the gold complex (SP,P)-[Helphos-K-1]AuCl, C1a (ORTEP = Oak Ridge Thermal-Ellipsoid Plot Program).
The ratios of endo/exo epimers depend mainly on the nature of the helical scaffold and the relative configurations of the stereogenic elements, going from 1:1, as in the reaction reported in Scheme 17, to >95:5 in favor of the endo-isomer, as observed notably for some phosphathiahelicene complexes [18c]. The phosphahelicene–gold complexes mentioned in this review are displayed in Table 2.
Phosphahelicene–gold complexes.
| Ligand | Gold complex | Ligand | Gold complex |
| Helphos-K-1 | Image 2 | Helphos-K-1 | Image 7 |
| L7 | Image 3 | Helphos-K-2 | Image 8 |
| Helphos-K-3 | Image 4 | Helphos-S-1 | Image 9 |
| Helphos-S-2 a, R = Ph b, R = 3,5-Me2C6H3 c, R = 4-Ph-C6H4 d, R = CC-Ph | Image 5 | Helphos-Y-1 | Image 10 |
| Helphos-P-1 | Image 6 |
3.2.1.2 Cycloisomerization of 1,6-enynes
In initial studies, the phosphahelicene–gold(I) complexes have been extensively evaluated as catalysts for the enantioselective cycloisomerization of N-tethered 1,6-enynes, such as 36, into 3-azabicyclo[4.1.0]heptenes, typified by the benchmark reaction in Scheme 18 [25]. The gold chloride precatalysts [L*AuCl] are activated by addition of an appropriate silver salt AgX (X = BF4, SbF6, NTf2, etc.), which converts the precatalyst into a cationic gold complex by removal of the chloride ligand.
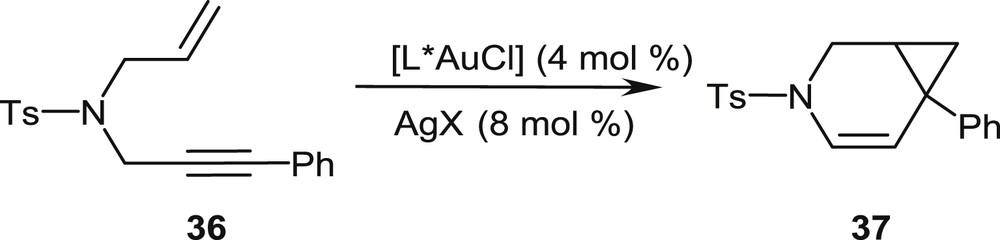
Gold-promoted cycloisomerization of an N-tethered enyne into a 3-azabicyclo[4.1.0]heptene.
These systematic studies have highlighted some general trends in terms of structure/activity and structure/enantioselectivity relationships (Fig. 5), which are summarized as follows. (1) At first, in terms of catalytic activity, the endo-isomers of the gold complexes usually display higher catalytic activity than the corresponding exo-isomers, which is probably related to a higher stability of the catalyst when gold is located into the cavity of the helical pocket. (2) In terms of enantioselectivity levels, higher enantiomeric excesses are obtained when the phosphorus atom is located on the internal rim of the helical scaffold. Thus, for instance, the [7]helicene in Fig. 6, where phosphorus lies on the external edge of the helical structure, gives much lower enantiomeric excesses than the corresponding [6]helicene shown in Fig. 5 (R = Ph). (3) Efficient enantiocontrol necessitates that the helical ligand contains an α-substituted phosphole ring (R ≠ H in Fig. 5). Notably, phenyl and substituted aryls are suitable substituents. (4) Endo-isomers of the gold complexes give higher enantioselectivity than the exo analogues. This behavior may be easily explained because the endo-configuration brings the metal center closer to the stereogenic helical structure. (5) Finally, diastereomers of the same complex with different relative configurations of the stereogenic elements (helical scaffold, phosphorus atom, and chiral phosphorus substituent) may give significantly different enantioselectivity levels and, therefore, the optimal, “matching” diastereomer must be sought.
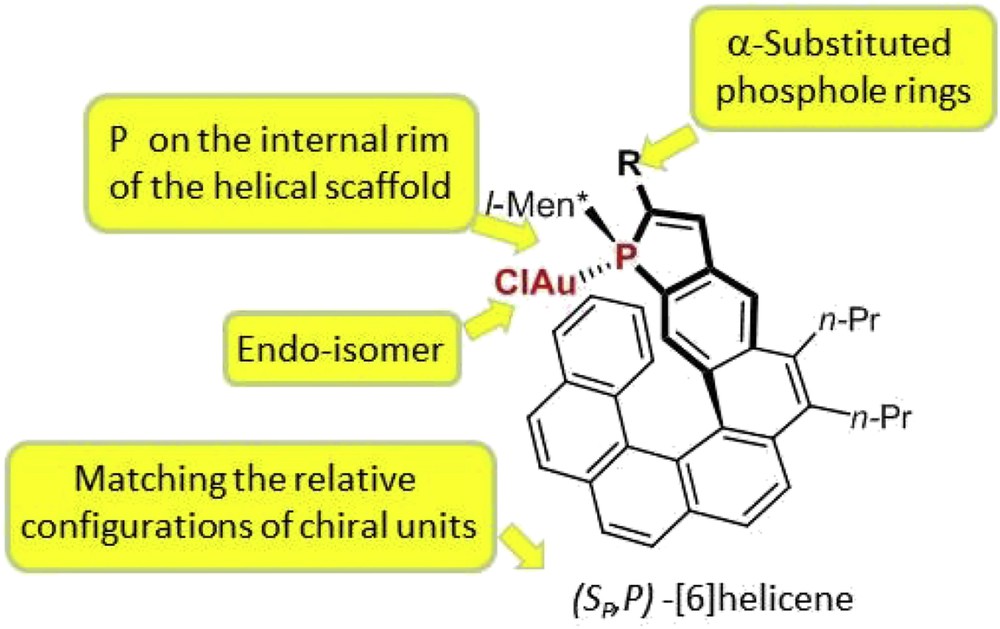
Structural requirements for high enantioselectivity levels.
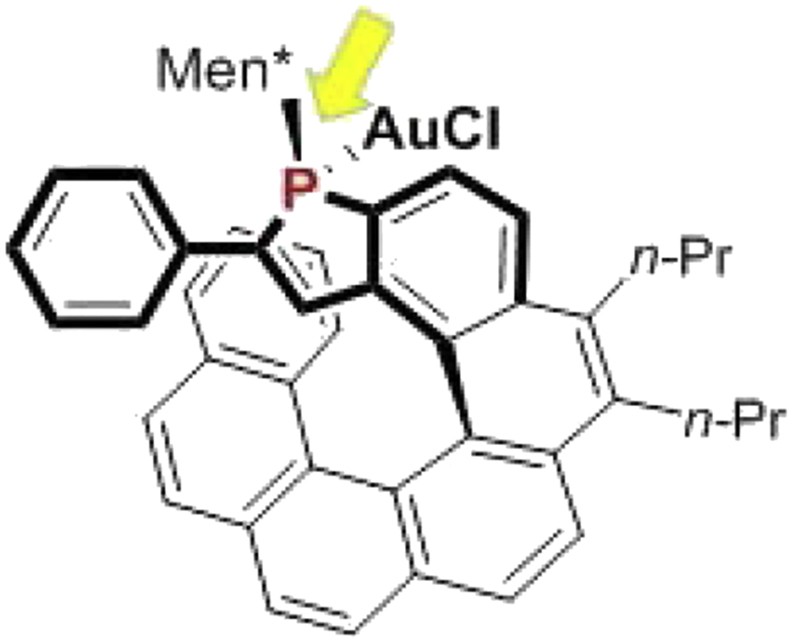
A [7]helicene displaying phosphorus on its external rim.
As a consequence of the aforementioned structural requirements (point (2)), catalytic studies have been focused so far mainly on phosphahelicenes in which the phosphole ring is meta-condensed to the helical sequence of aromatic rings, the so-called Helphos series. Then, within homogeneous series of Helphos ligands, with a given helical scaffold, the substitution patterns and relative configurations of the stereogenic centers have been optimized.
Representative results of the catalytic tests are displayed in Table 3. The highest enantioselectivity was attained with a ligand of the Helphos-K series, giving an 84% ee and full conversion at room temperature after 24 h. The best ligand, Helphos-K-2, displays a bulky 3,5-dimethylphenyl substituent on the phosphole ring, α-to the phosphorus atom. The analogous phenyl-substituted ligand affords a slightly lower enantiomeric excess, 81% ee.
Screening of phosphahelicene–gold complexes in the cycoisomerization of an N-tethered 1,6-enyne.
| Complex | Ligand | Yield (%) | ee (%) (configuration) | Reference |
| C2a | L7 | 35 | 43 | [25] |
| C1b | Helphos-K-1 | <5 | – | [25] |
| C1a | Helphos-K-1 | >95 | 81 | [25] |
| C3a | Helphos-K-2 | >95 | 84 (1R,6S) | [25] |
| C4a | Helphos-K-3 | >95 | 82 | [25] |
| C5a | Helphos-S-1 | 78 | 74 | [18c] |
| C7a | Helphos-Y-1 | 88 | 24 | [b] |
In further studies, the catalytic uses of the same gold complexes have been extended to the cycloisomerization of N-tethered 1,6-enynes 38 displaying an additional olefin function conjugated to the alkyne unit (Table 4). The outcome of these reactions depends on the substitution pattern of the olefinic unit of the enyne: substrate 38a displaying a monosubstituted olefin (R = H) affords the expected bicyclo[4.1.0]heptene 39, whereas for R = Ph (38b) the intermediate bicyclo[4.1.0]heptene undergoes a stereocontrolled vinylcyclopropane–cyclopentene rearrangement leading to the tricyclic compound 40 as a single diastereomer. In both reactions, the Helphos-S-1 ligand afforded very high enantiomeric excesses (89% and 96% ee, respectively), when the gold chloride precatalyst was activated by addition of silver triflimide.
Helphos-catalyzed cycloisomerizations of 1-cyclohexenyl-substituted 1,6-enynes.
| Complex | Ligand | AgX | Product | Yield (%) | ee (%) | Reference |
| C1a | Helphos-K-1 | AgBF4 | 39 | 70 | 86 | [25] |
| C5a | Helphos-S-1 | AgNTf2 | 39 | 87 | 89 | [18c] |
| C1a | Helphos-K-1 | AgBF4 | 40 | 60 | 84 | [25] |
| C4a | Helphos-K-3 | AgBF4 | 40 | 50 | 85 | [25] |
| C5a | Helphos-S-1 | AgBF4 | 40 | 50 | 65 | [18c] |
| C5a | Helphos-S-1 | AgNTf2 | 40 | 53 | 96 | [18c] |
Then, in a new series of experiments, the substrates have been varied by considering the N-tethered enyne 41 in which the olefin function is embedded into a cyclic unit (Scheme 19). The expected cycloisomerization process afforded the tricyclic compound 42 including a cyclopropane unit in its core structure. A 93% ee was obtained with the gold triflimidate of Helphos-S-1, when the reaction was carried out at 0 °C [18c].

Enantioselective cycloisomerization of a 1,6-enyne displaying a cyclic alkene moiety.
In all reactions mentioned previously, the key step is postulated to be the electrophilic activation of the alkyne unit through π-complexation by the cationic gold complex, followed by the intramolecular attack of the alkyne by the nucleophilic olefin moiety (Fig. 7). This is expected to be the stereodetermining step. On the basis of this hypothesis, a stereochemical model has been proposed to account for the observed stereoselectivity. It is displayed in Fig. 7 for the cycloisomerization of the model N-tethered 1,6-enyne 36 with the Helphos-K-1 complex C1a. The X-ray crystal structure of the precatalyst C1a (see Fig. 4) indicates that three space regions around gold are hindered, respectively, by the phenyl substituent on the phosphole ring (upper-left quadrant), by the menthyl group (upper-right quadrant), and by the folded helical scaffold itself (bottom-right quadrant). During the cyclization step, the remaining space region, that is, the bottom-left quadrant, is expected to accommodate the group with the highest steric hindrance. The most hindered group is postulated to be the phenyl substituent of the alkyne unit.
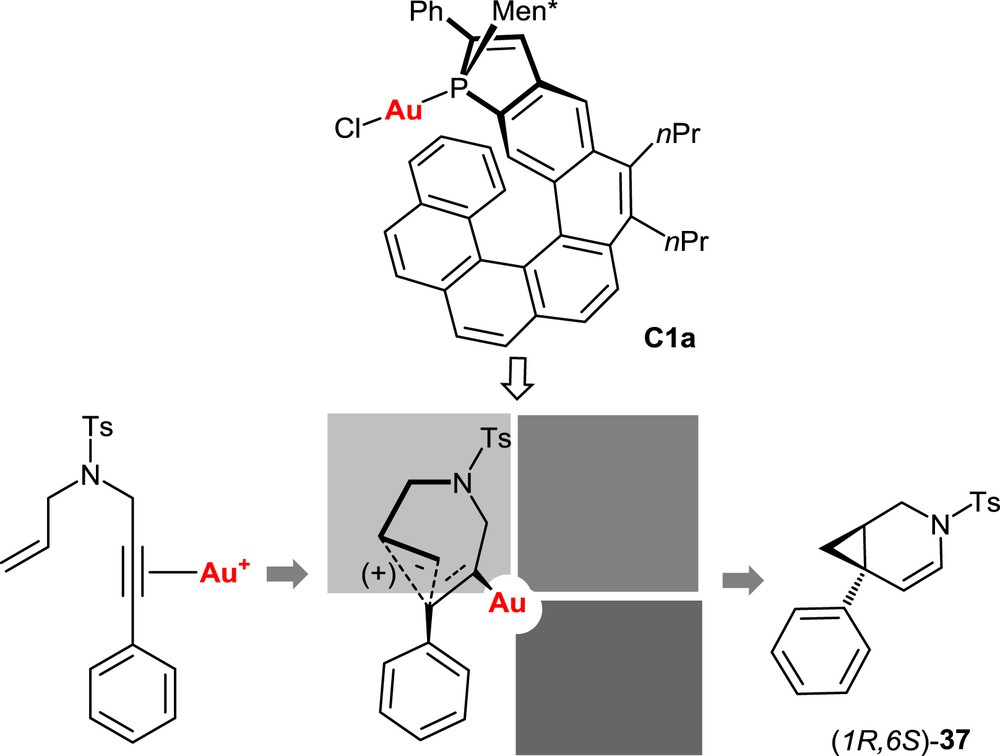
Stereochemical model for the cycloisomerization of the N-tethered 1,6-enyne 36 promoted by the Helphos-K-1 complex C1a.
This simple model suitably accounts for the observed stereochemical outcome of the reaction, namely the preferential formation of the (1R,6S)-configured bicyclo[4.1.0]heptene 37.
Further catalytic studies have been devoted to the cycloisomerization of the 1,6-enyne 43 displaying a carbon tether between the alkyne and the alkene functions (Table 5) [20]. In the presence of metal catalysts this substrate undergoes a formal [4+2] cycloaddition involving both the enyne moiety and one of the double bonds of the phenyl substituent of the alkyne, leading to 44 [27]. This reaction has served notably to evaluate the effects of the substitution pattern in Helphos-S ligands. Indeed, as already mentioned in Section 3.1.1 (Scheme 15), the terminal thiophene unit of these ligands can be functionalized easily through regioselective procedures and various substituents can be introduced on the α- to sulfur position. The catalytic tests have demonstrated that the nature of the thiophene substituents tunes significantly the enantiomeric excess. In this reaction, the highest enantioselectivity was obtained with Helphos-S-2d for which R = CCPh.
Cycloisomerization of a 1,6-enyne via formal [4+2] cycloaddition, promoted by gold complexes of Helphos-S.
| Complex | Ligand | R | T | Yield (%) | ee (%) |
| C5a | Helphos-S-1 | H | rt | 95 | 70 |
| C5a | Helphos-S-1 | H | −20 °C | 92 | 81 |
| C6aa | Helphos-S-2a | Ph | rt | 99 | 84 |
| C6ab | Helphos-S-2b | 3,5-Me2C6H3 | rt | 80 | 80 |
| C6ac | Helphos-S-2c | 4-Ph-C6H4 | rt | 81 | 82 |
| C6ad | Helphos-S-2d | CCPh | rt | 99 | 91 |
Although the observed substituent effects cannot be rationalized easily, this work discloses that variation in these distal substituents of the ligand allows an efficient optimization of the catalyst. In this regard, the direct and easy, late functionalization of the preformed helicene scaffold in Scheme 15 represents a highly valuable synthetic tool.
3.2.1.3 Cycloisomerization of 1,6-allenenes and tandem reactions
Beyond enynes, a number of polyunsaturated substrates are known to undergo cycloisomerization under transition metal catalysis. Among them, allenenes represent highly versatile substrates, which rearrange into a variety of synthetically valuable polycyclic scaffolds [28]. Only a few enantioselective variants of these processes have been reported so far under gold catalysis. In this context, the Helphos gold complexes have been evaluated as catalysts for some of these reactions, namely the [2+2] and [4+2] cyclizations [29] of the 1,6-allenenes shown in Table 6 [22b].
Enantioselective cycloisomerizations of 1,6-allenenes.
| Entry | Ligand (complex) | X | Product | Solvent | T | Conversion (%) | ee (%) |
| 1 | Helphos-K-1 (C1a) | NTs | 46a | Toluene | rt | >95 | 76 |
| 2 | Helphos-S-1 (C5a) | NTs | 46a | Toluene | rt | >95 | 58 |
| 3 | Helphos-P-1 (C8a) | NTs | 46a | Toluene | rt | >95 | 79 |
| 4 | Helphos-P-1 (C8a) | NTs | 46a | PhNO2 | 6 °C | >95 | 88 |
| 5 | Helphos-P-1 (C8a) | C(SO2Ph)2 | 46b | (ClCH2)2 | rt | 85% Yield | 92 |
| 6 | Helphos-P-1 (C8a) | C(SO2Ph)2 | 47 | (ClCH2)2 | rt | 83% Yield | 84 |
Two series of substrates have been considered: a 1,6-allenene with a nitrogen-containing tether (entries 1–4) and a 1,6-allenene displaying an all-carbon tether in which the central carbon bears sulfonyl functions (entries 5 and 6). Substrates with aryl-substituted olefin functions undergo [2+2] cyclizations leading to 46. In these reactions, the benzo-fused Helphos-P-1 catalyst gave high conversion rates, in mild conditions, and enantiomeric excesses up to 88% and 92% for the two substrates respectively (entries 4 and 5). When the substrate displays a diene function, instead of a simple olefin function (R = CHCH2), the [4+2] cyclization product 47 is formed in 84% ee, by using the same catalyst. These enantiomeric excesses are the highest reported so far for the [2+2] and [4+2] cyclizations of sulfonyl-functionalized substrates.
The sulfonyl-functionalized allenenes 48 have been submitted also to tandem cyclization/nucleophilic addition reactions. In the presence of water or alcohols, the gold-promoted cycloisomerization process stops after formation of the first CC bond, because the nucleophilic agent traps one of the carbocationic intermediates. Thus, the reaction affords the substituted cyclopentanes 49 displaying ether or alcohol functions (Scheme 20). These products were isolated in high enantiomeric excesses when the Helphos-P-1 gold complex C8a was used as the catalyst (ee up to 90%) [22b].
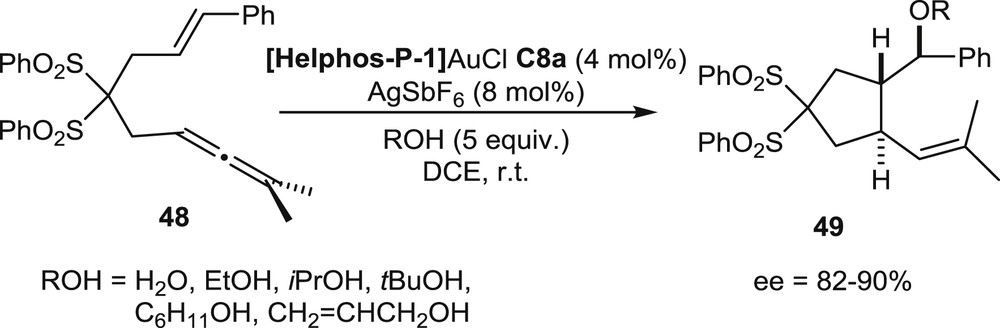
Tandem cyclization/additions of oxygen nucleophiles.
On the whole, the reactions mentioned previously demonstrate that phosphahelicenes are well-suited ligands for enantioselective gold catalysis. They have the potential to complement the previous catalytic systems and expand their catalytic scope.
3.2.2 Nucleophilic organocatalysis
In the field of phosphine organocatalysis, several recent studies have been devoted to enantioselective [3+2] cyclization reactions between electron-poor allenes and olefins leading to synthetically useful, highly functionalized cyclopentenes [24,30]. These reactions involve activation of the allene by nucleophilic addition of the trivalent phosphine. Phosphahelicenes bearing either an l-menthyl (Men*) or an isopinocampheyl (iPc*) group on phosphorus (Fig. 8) have been evaluated as catalysts for reactions of this class.
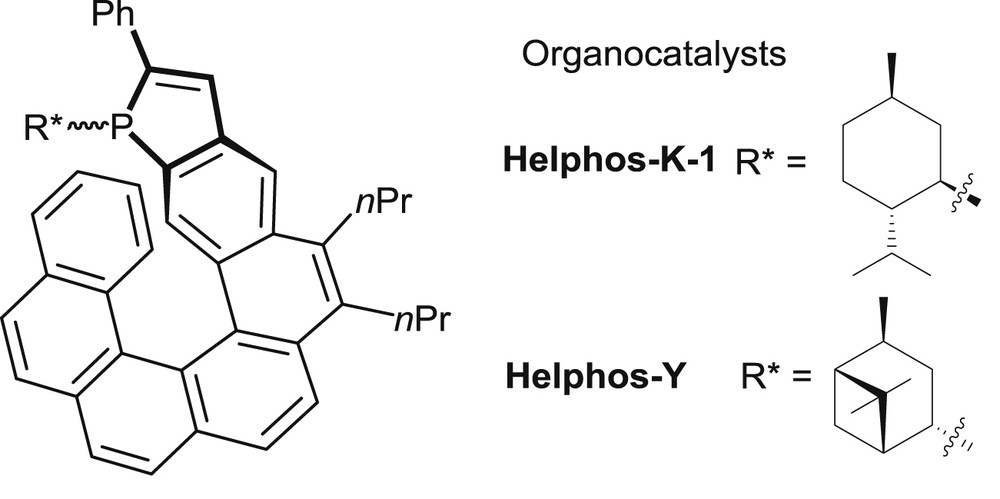
Trivalent phosphahelicenes tested in organocatalytic [3+2] cyclizations.
The main results are summarized in Table 7 [19]. In the cyclization between a γ-substituted allenoate (R2 = CH2CH2Ph) and 1,1-dicyano-2-phenylethylene both phosphines displayed excellent enantioselectivity, the iPc* group giving a slightly better enantiomeric excess (89% vs 95% ee, entries 1 and 2). Conversion rates were moderate at 60 °C, but could be improved in reactions performed at 80 °C, without loss of enantioselectivity (entry 3). In the P-iPc*-substituted Helphos-Y series, the catalyst with (P)-configuration of the helical scaffold, displayed in Fig. 8, performs better than the corresponding (M)-configured catalyst. Thus, the (P)-configured Helphos-Y catalyst was screened extensively in the [3+2] cyclizations of variously substituted dicyanoalkenes 50 and γ-substituted allenoates 51. It afforded fully satisfying yields and diastereoselectivity, as well as very high enantioselectivity levels (19 examples, ee = 82–96%).
Organocatalytic enantioselective [3+2] cyclizations.
| Entry | Catalyst | R1 | R2 | T | Yield (%) | d.r. | ee(%) |
| 1 | Helphos-K-1 | Ph | (CH2)2Ph | 60 °C | 30 | >95:5 | 89 |
| 2 | Helphos-Y | Ph | (CH2)2Ph | 60 °C | 37 | >95:5 | 95 |
| 3 | Helphos-Y | Ph | (CH2)2Ph | 80 °C | 91 | >95:5 | 96 |
| 4 | Helphos-Y | 4-ClC6H4 | (CH2)2Ph | 80 °C | 84 | 95:5 | 94 |
| 5 | Helphos-Y | 4-MeOC6H4 | (CH2)2Ph | 80 °C | 84 | >95:5 | 94 |
| 6 | Helphos-Y | 2-BrC6H4 | (CH2)2Ph | 80 °C | 94 | >95:5 | 87 |
| 7 | Helphos-Y | 2-Thienyl | (CH2)2Ph | 80 °C | 80 | 90:10 | 95 |
| 8 | Helphos-Y | Ph | CH2C5H9 | 80 °C | 86 | >95:5 | 95 |
| 9 | Helphos-Y | Ph | CH3 | 80 °C | 73 | 95:5 | 94 |
The excellent stereocontrol induced by Helphos-Y is especially remarkable because the phosphorus center of the trivalent phosphine is known to be configurationally unstable both at 80 °C and at room temperature. This means that the catalyst here should be an epimeric, equilibrating mixture of phosphines with opposite configurations at phosphorus. NMR monitoring has shown indeed that, starting from a single epimer of the trivalent phosphine, a 2:3 ratio of epimers is reached at 60 °C in toluene. At this time, mechanistic studies are needed to enlighten the precise nature and behavior of these catalysts.
Finally, the efficiency of Helphos-Y as an organocatalyst was also demonstrated in the [3+2] cyclizations of cyanoallenes 53 with 2-arylidenes-malononitriles 50 shown in Scheme 21.

Organocatalyzed [3+2] cyclizations on buta-2,3-dienenitriles.
Cyanoallenes display higher reactivity than allenoates and, therefore, these cyclization reactions were carried out at room temperature. In the presence of Helphos-Y, the desired cyclopentene was obtained with total diastereoselectivity and over 80% ee. These are the first examples of highly enantioselective phosphine-promoted cyclizations on cyanoallenes reported to date.
4 Conclusions
The exhaustive review of the catalytic uses of phosphorus-containing helicenes as ligands or organocatalysts highlights four main points: (1) despite the multitude of known homogeneous organometallic catalytic reactions, very few of them have been investigated with helical ligands with appended phosphorus functions. Mainly benchmark palladium- or iridium-promoted allylic substitutions and rhodium-promoted hydrogenations and hydroformylations have been considered. (2) The enantiomeric excesses are often moderate; however, appropriate design allows good enantiomeric excesses to be attained in a few instances, such as in iridium-promoted allylic aminations and palladium-promoted Suzuki–Miyaura couplings. (3) The recent development of phosphahelicenes has afforded significant advances in the challenging field of enantioselective gold catalysis. In this case, diverse and versatile synthetic approaches allow proper design, fine tuning of the backbones, and substitution patterns of the ligands, which are crucial for the optimization of the catalytic performances. (4) Finally, in the field of organocatalysis, suitably designed phosphahelicenes have afforded highly enantioselective catalysts for [3+2] cyclization reactions.
Although many initial studies have been focused on helicenes with phosphorus functions on their external faces, the most recent advances in asymmetric catalysis result from ligands/organocatalysts in which the phosphorus function is oriented toward the internal face of the helical pocket. An analogous design has been developed also for pyridine-based organocatalysts [31]. All these ligands/organocatalysts fully benefit from the three-dimensional chiral environment created by the helical backbone itself to generate highly effective chiral induction. It can be easily anticipated that this new design will be further applied in the future not only to the synthesis of new chiral phosphorus auxiliaries but also to other families of carbon or nitrogen-based ligands and organocatalysts.
Acknowledgments
The authors would like to thank the ANR agency for grant to C.S.D. in the frame of the “Helphos” project (ANR-15-CE29-0012) and CEFIPRA for financial support (Project No. 5505-2, 2016).